




Clinical record
A 50‐year‐old man presented with one week of progressive dyspnoea, abdominal pain and malaise, together with one month of enlarging capillary haemangiomas. His medical history reported post‐coronavirus disease 2019 (COVID‐19) multisystem inflammatory syndrome in adults (MIS‐A), diagnosed 18 months prior with dyspnoea and abdominal pain, and associated with progressive kidney injury, liver function derangements, pancytopenias and mediastinal lymphadenopathy. MIS‐A was diagnosed clinically after non‐diagnostic mediastinal lymph node and liver biopsies. He was treated with corticosteroids, which were weaned only five months before the current presentation.
On presentation, his vital signs and initial examination were unremarkable. Blood results showed anaemia, inflammation, acute kidney injury and cholestatic liver derangement (Box 1). A chest x‐ray showed small bilateral pleural effusions, further evaluated with computed tomography pulmonary angiography, which revealed mediastinal and axillary adenopathy. Serological workup revealed unremarkable infectious, autoimmune and haematologic screens (Box 2).
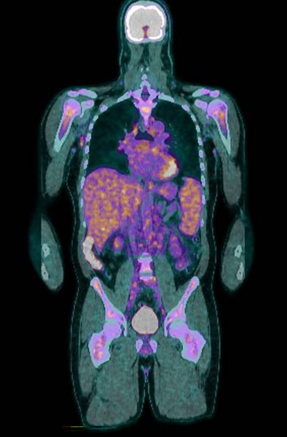
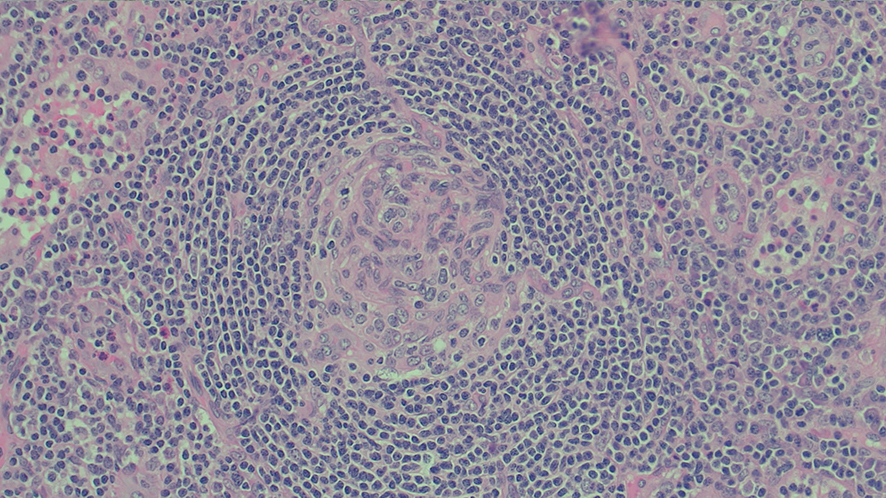
A positron emission tomography scan showed innumerable mildly to moderately fludeoxyglucose (FDG)‐avid enlarged and non‐enlarged lymph nodes above and below the diaphragm, and increased splenic and bone marrow activity (Box 3). Biopsy samples were taken from the most FDG‐avid axillary and mediastinal lymph nodes, but were non‐diagnostic. The results from one biopsy showed non‐caseating granulomas; however, results from the second biopsy did not. An excisional lymph node biopsy was subsequently taken, with the results meeting histopathologic criteria for Castleman disease (Box 4). These criteria included depleted germinal centres, concentric rimming of small B lymphocytes in a “onion skin” appearance, and vascularity with vessels extending into the germinal centre.1
The patient was diagnosed with multicentric Castleman disease, and commenced on high dose prednisolone and interleukin‐6 inhibitor siltuximab. At the time of writing, he was in incomplete remission.
Discussion
The 50‐year‐old‐patient presented with exuberant multisystem inflammation. With a history of MIS‐A, a relapse was initially considered; however, there are no documented cases of relapsed MIS‐A. Thus, alternative causes were considered, especially in the context of extensive lymphadenopathy.
Core lymph node biopsy results revealed non‐caseating granulomas, initially suspicious of sarcoidosis. However, non‐caseating granulomas may also indicate many viral, bacterial or fungal infections, vasculitides, occupational diseases and haematological disorders.2 Core lymph node biopsies can provide definitive diagnosis in more than 92% of cases. However, they have an increased rate of incorrect and non‐conclusive diagnosis compared to an excisional lymph node biopsy.3 Excisional lymph node biopsies should be pursued if any diagnostic ambiguity exists. In this case, excisional lymph node biopsy results led to our diagnosis of Castleman disease.
Castleman disease is a rare lymphoproliferative disorder that can present with a solitary enlarged lymph node (unicentric Castleman disease) or multiple enlarged lymph nodes (multicentric Castleman disease; MCD). Most cases are idiopathic, with rarer subtypes associated with human herpesvirus‐8, and the neoplastic POEMS syndrome (polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes).2
The diagnosis of idiopathic MCD requires the presence of two major criteria, and at least two of 11 minor criteria, including clinical and laboratory criteria. The major criterion was: two enlarged lymph nodes and a lymph node meeting histopathological criteria for MCD. Minor criteria include eruptive cherry angiomatosis or violaceous papules. Other minor clinical criteria include constitutional symptoms, hepatomegaly or splenomegaly, fluid accumulation, and lymphocytic interstitial pneumonitis. Minor laboratory criteria include elevated inflammatory markers, anaemia, thrombocytopenia or thrombocytosis, hypoalbuminemia, renal dysfunction or proteinuria, and polyclonal hypergammaglobulinemia.2
First‐line treatment is with interleukin‐6 blockade, typically siltuximab, accompanied by high dose prednisolone. Second‐line therapy for MCD includes rituximab, corticosteroids, thalidomide, ciclosporin, sirolimus, traditional chemotherapy and stem cell transplant.2
The patient's first symptom of cherry haemangioma enlargement was a clue to the aetiology. Eruptive cherry haemangiomas are suspected to be driven by vascular endothelial growth factor, one of the early cytokines released in the cytokine storm from MCD.1
However, the main cytokine driving MCD is thought to be interleukin‐6, the same major cytokine driving severe COVID‐19 infections and MIS‐A.2,4 The shared interleukin‐6 driver explains similarities in their presentations, raises the biological plausibility that MIS‐A could evolve into Castleman disease, and cautions that MIS‐A could be masquerading as Castleman disease.
This article adds to the literature suggesting a possible causal link between COVID‐19 and Castleman disease, with growing case reports identifying Castleman disease cases after COVID‐19 infection and COVID‐19 vaccination.5 To our knowledge, this is the first case report of Castleman disease following post‐COVID‐19 MIS‐A.
Lessons from practice
- Consider Castleman disease in patients re‐presenting with similar symptoms as post‐coronavirus disease 2019 multisystem inflammatory syndrome in adults.
- Non‐caseating granulomas have a wide range of differentials; including infections, vasculitis, occupational diseases and haematological aetiologies.
- Excisional lymph node biopsy is the gold standard to diagnose unexplained lymphadenopathy.
- Enlargement of cherry angiomas is a sign of Castleman disease.
Box 1 – Laboratory values
|
Laboratory value |
Value |
Reference range |
|||||||||||||
|
|
|||||||||||||||
|
Haemoglobin (g/L) |
127 |
135–175 |
|||||||||||||
|
White cell count (109/L) |
8.67 |
4–11 |
|||||||||||||
|
Platelet count (109/L) |
173 |
150–450 |
|||||||||||||
|
C‐reactive protein (mg/L) |
109.7 |
0–8 |
|||||||||||||
|
Creatinine (μmol/L) |
111 |
60–110 |
|||||||||||||
|
Estimated glomerular filtration rate (mL/min/1.73m2) |
66 |
≥ 90 |
|||||||||||||
|
Albumin (g/L) |
32 |
34–48 |
|||||||||||||
|
Aspartate transaminase (U/L) |
28 |
0–45 |
|||||||||||||
|
Alanine aminotransferase (U/L) |
16 |
0–55 |
|||||||||||||
|
Alkaline phosphatase (U/L) |
240 |
30–110 |
|||||||||||||
|
Gamma‐glutamyl transferase (U/L) |
231 |
0–60 |
|||||||||||||
|
Bilirubin (μmol/L) |
6 |
2–24 |
|||||||||||||
|
Angiotensin‐converting enzyme (U/L) |
< 7 |
20–70 |
|||||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Box 2 – Screening tests ordered
|
Screening test categories |
Screening tests ordered |
||||||||||||||
|
|
|||||||||||||||
|
Infectious tests |
Blood cultures, urine cultures, viral nasopharyngeal polymerase chain reaction, hepatitis serology, Epstein–Barr virus, cytomegalovirus, human immunodeficiency virus |
||||||||||||||
|
Autoimmune tests |
Antinuclear antibody, anti‐double‐stranded deoxyribonucleic acid antibodies, extractable nuclear antigen, antineutrophil cytoplasmic antibodies, immunoglobulin G4, complements 3 and 4 |
||||||||||||||
|
Haematological tests |
Flow cytometry, serum protein electrophoresis, serum free light chains |
||||||||||||||
|
|
|||||||||||||||
|
|
|||||||||||||||
Provenance: Not commissioned; externally peer reviewed.
- 1. Judson MA. Granulomatous sarcoidosis mimics. Front Med (Lausanne) 2021; 8: 680989.
- 2. Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence‐based consensus diagnostic criteria for HHV‐8–negative/idiopathic multicentric Castleman disease. Blood 2017; 129: 1646‐1657.
- 3. Surukh C, Chaouat C, Poullott E, et al. Lymph node excisions provide more precise lymphoma diagnoses than core biopsies: a French Lymphopath network survey. Blood 2022; 140: 2573‐2583.
- 4. Patel P, DeCuir J, Abrams J, et al. Clinical characteristics of multisystem inflammatory syndrome in adults. JAMA Netw Open 2021; 4: e2126456.
- 5. Liu Y, Yin X, Xu D, et al. When idiopathic multicentric Castleman disease meets COVID‐19: a multicenter retrospective study from China. Ther Adv Hematol 2024; 15: 20406207241229584.
Patient consent:
The patient provided written consent for publication.
No relevant disclosures.