





Cardiovascular disease (CVD) is the leading cause of morbidity and mortality globally and is responsible for an estimated 32% of deaths.1 An effort over many decades targeting modifiable risk factors has resulted in significant improvements in mortality and age‐adjusted prevalence. However, the improved medical management and decreased mortality have also resulted in a 193% increase in overall CVD prevalence globally over the past 30 years. New approaches to prevention and early disease detection are greatly needed. Genetic testing has been incorporated into routine management of monogenic CVD, such as hypertrophic cardiomyopathy and primary arrhythmic syndromes. Advances in sequencing and computational biology now additionally provide unprecedented insights into the genetic contribution to common complex conditions, including coronary artery disease (CAD) and atrial fibrillation. Maximising the use of these genetic tools for the identification and prediction of CVD will require innovative prospective implementation studies and a rapid upskilling of our health workforce beyond the traditional clinical genetic pathways.
This narrative review seeks to provide an overview of the current evidence and recommendations for genetic testing in CVD, as well as evidence behind emerging platforms (eg, polygenic risk scoring). We searched the online databases PubMed and MEDLINE between September 2022 and April 2023 for expert consensus guidelines, consensus opinions, systematic reviews, meta‐analyses and review manuscripts in English language published since 1 January 2010. Search terms included “cardiovascular disease[MESH Major Topic]” or “cardiovascular diseases[MESH Major Topic]” and/or “genetic test”, “genetic risk”, “genetic risk score”, “polygenic risk score”, “genome‐wide association study”, and “guidelines”. Manuscripts included in these documents were individually reviewed and included as appropriate based on authors’ experience.
Monogenic conditions
Monogenic disorders with Mendelian inheritance patterns are characterised by the presence of a single genetic variant with large effect size that is sufficient to lead to a disease phenotype (Box). Monogenic diseases can have dominant, recessive or x‐linked inheritance patterns, and are categorised according to their autosomal or mitochondrial gene location. Despite well characterised examples in the CVD space, spanning from severe derangements in lipid metabolism to myocardial conditions associated with arrhythmias and early sudden cardiac death, these make up only a small proportion of the burden of CVD. Some important examples are described below based on inclusion in expert consensus guidelines.
Lipid metabolism
Familial hypercholesterolaemia is an autosomal dominant condition characterised by significantly elevated concentrations of circulating low‐density lipoprotein (LDL) cholesterol (LDL‐C) and is the most common monogenic condition in humans, affecting an estimated one in 300 people.2 Nearly 1500 pathogenic variants have been identified across the three main causative genes (LDLR, APOB and PCSK9) affecting components of plasma lipoprotein cellular uptake and these collectively have a population prevalence of 0.32%.3 Case–control studies have demonstrated a nearly tenfold increased familial hypercholesterolaemia prevalence in people with CAD and more than 20‐fold increased prevalence in people with premature CAD compared with the general population.3 Familial hypercholesterolaemia is most frequently diagnosed clinically using the Dutch Lipid Clinic Network and/or Simon Broome tools.4,5 However, these clinical diagnostic algorithms have limitations, particularly when diagnosing children and adolescents. Identification of pathogenic variants can facilitate cascade testing, allowing for earlier diagnosis and aggressive LDL‐targeting pharmacotherapy for biological family members, thus having a substantial impact on medical management.
Thoracic aortic disease
Heritable thoracic aortic disease is defined as aortic aneurysms and/or dissection with strong genetic association and no other discernible features. This, therefore, often results in catastrophic clinical presentation, as only about 5% of patients with thoracic aortic aneurysm and dissection are symptomatic before dissection.6 The lack of physical features results in difficulty in assessing true prevalence and heritability, with a predicted incidence of 2.9–4.3 cases per 100 000 person‐years and suspected autosomal dominant inheritance pattern.7,8 Eleven genes have been identified as definitively or strongly associated with heritable thoracic aortic disease, with most of them relevant to maintaining vascular wall integrity. The role of vascular smooth muscle cell dysfunction in the development of thoracic aneurysms has been well characterised,9 and, indeed, these genes can be grouped based on the role of the coded protein in smooth muscle cell contractile function, extracellular matrix adhesion, and transforming growth factor‐β (TGF‐β) signalling.10 A variant in ACTA2, encoding α‐2 actin with smooth muscle cell‐specific contractile function, is the most common genetic variant associated with heritable thoracic aortic disease, with a penetrance of about 50% and identified in 12–21% of cases.11,12 Genetic screening programs have been proposed due to their potential impact on surgical intervention and familial case identification, especially in patients with early onset, a positive family history of dissection, and no history of hypertension.13
In addition, causative pathogenic gene variants have been identified in syndromes with vascular manifestations; for example, vascular Ehlers–Danlos (COL3A1),14 Loeys–Dietz (TGFBR1/2),15,16 and Marfan (FBN1) syndromes.17,18 Genetic testing in these settings aids identification of the syndrome and can play an important role, as there are substantial differences in how the vasculature is affected in each syndrome. Genes associated with vasculopathies, including COL3A1 and TGFBR2, have additionally been implicated in other forms of arterial dissection, including spontaneous coronary artery dissection.19,20
Inherited cardiomyopathies
Inherited cardiomyopathies are a spectrum of cardiac diseases broadly classified into hypertrophic, dilated, arrhythmogenic and restrictive phenotypic subtypes, which are not explained by the presence of ischaemic, congenital, hypertensive or valvular disease. Although epidemiological data are scarce due to underdiagnosis, at least 0.6% of the population are estimated to be affected by this spectrum of diseases, with significant differences in clinical presentation depending on sex and race.21
Hypertrophic cardiomyopathy is a primary myocardial disorder that is characterised by asymmetric ventricular hypertrophy and distinctive histopathological features, including myofibril hypertrophy, disarray, and interstitial fibrosis. Hypertrophic cardiomyopathy is generally caused by deleterious variants in sarcomere genes, with variants in MYH7 and MYBPC3 identified in nearly 70% of cases.22 It is important to differentiate hypertrophic cardiomyopathy from other heritable disorders that mimic this condition, some of which have specific therapies, including glycogen storage disorders, Fabry disease and cardiac amyloidosis.23 Despite decades of research into the genetic underpinnings of hypertrophic cardiomyopathy, a subset of patients will have negative results on genetic testing. Although this may reflect undiscovered monogenic forms of disease, it has been proposed that ventricular hypertrophy, in some cases, might result from combinations of common variants.
Dilated cardiomyopathy is a disorder with prevalence estimates of up to one in 250 persons and has numerous causes, which can be genetic or acquired.24,25 These distinctions are increasingly blurred, with evidence of rare genetic variants in patients with environmental causes of dilated cardiomyopathy and vice versa. A genetic basis for disease is often not apparent in the index case, and a positive family history may only emerge after successive relatives are diagnosed.26 Although more than 250 disease genes have been reported, recent gene curation efforts have shown that only ten to 20 genes currently have robust evidence for dilated cardiomyopathy causation (Supporting Information, table 1).26,27,28 Truncating variants in the gene TTN (TTNtv), which encodes the sarcomeric protein titin, are the most common genetic cause of dilated cardiomyopathy, present in up to 20% of cases.29 Interpreting the clinical significance of TTNtv may be challenging, as these variants are also present in up to 3% of the general population.30 It has been proposed that a “second hit” might modify severity in TTNtv carriers, which is currently an area of active research.31,32
Arrhythmogenic cardiomyopathy is an umbrella term that refers to a group of highly arrhythmic cardiomyopathies that variably affect the right and/or left ventricles.33 This includes arrhythmogenic right ventricular cardiomyopathy that mainly affects the right ventricle, but frequently has left ventricular involvement, and arrhythmic forms of dilated cardiomyopathy, such as those associated with LMNA, FLNC, RBM20 and SCN5A variants.34 Arrhythmogenic right ventricular cardiomyopathy exhibits autosomal dominant inheritance in most cases, with pathogenic variants identified in desmosomal genes.35 There is often incomplete penetrance with variable expressivity. Truncating variants in PKP2 are most frequently implicated and typically give rise to the right‐sided phenotype.36 In addition, pathogenic variants have been identified in non‐desmosome encoding genes, albeit all with less frequency.
Inherited arrhythmic disorders
Inherited arrhythmic syndromes include monogenic conditions such as long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, and familial forms of atrial fibrillation. LQTS is one of the most common inherited cardiac conduction diseases, which can lead to sudden cardiac death and is believed to occur in about 50 infants per 100 000.37 Nearly 80% of patients with a prolonged corrected QT interval, in the absence of structural heart disease and exposure to QT prolonging drugs, have an identifiable LQTS‐associated genetic variant,38 which mostly demonstrates an autosomal dominant inheritance pattern. Three genes have been definitively associated with LQTS. LQT1 and LQT2 have been associated with potassium channel protein‐encoding genes KCNQ1 and KCNH2 respectively.39,40 Genetic association with LQT3 is more complex, with gain‐of‐function variants in SCN5A, which encode a sodium channel subunit, associated with disease and identified in about 10% of LQTS cases.41 Mutations in SCN5A have additionally been associated with Brugada syndrome, cardiac conduction abnormalities and dilated cardiomyopathy.
Complex genetics and cardiovascular disease
Advances in sequencing, computational power, analytical approaches, and global collaborations have been central to unlocking the contributions of genetic variation to commonly occurring complex CVD, such as CAD and atrial fibrillation.
Genome‐wide association studies (GWAS) using large case‐control studies have identified common variants associated with disease without surveillance bias to specific biological pathways or genes of interest. Due to the substantial overlap between cases and controls, the prognostic significance of a single‐nucleotide polymorphism (SNP) in an individual patient is limited. Polygenic risk scores (PRS) have been derived to increase the clinical utility of these data by assessing the collective effects of SNP profiles in individual subjects. Emerging research in certain complex CVD suggests that patients with very high scores have an increased risk of disease several‐fold higher than that of the population average, and indeed a risk level equivalent to risk conferred by some monogenic disorders.
CAD heritability is classically estimated to be about 40–60%,42,43 a proportion not fully explained by causal rare variants in genes associated with monogenic lipid disorders. More than 200 significant GWAS loci have been identified in European populations and incorporated into multiple CAD‐PRS algorithms. Only about half of included SNPs have previously been associated with traditional CAD risk factors, including lipid metabolism and blood pressure. In a pivotal study in 2016 that included data from more than 55 000 participants from four separate cohorts, the relative risk of incident CAD events was increased by 91% in participants with a top quintile CAD‐PRS compared with those in the bottom quintile.44 Importantly, the detrimental effect of a high CAD‐PRS was able to be offset by adopting a healthier lifestyle, with an observed 46% relative risk reduction of CAD events. In another study using CAD‐PRS of 1.7 million genetic variants, the relative risk of CAD was 4.17‐fold higher in participants with top versus bottom quintile scores, a result independent of traditional risk factors.45
Atrial fibrillation is the most common cardiac arrhythmia in Australia and affects about one in 18 Australians aged over 55 years.46 It confers an increased risk of stroke and heart failure and constitutes a growing health care and economic burden. Although atrial fibrillation can occur as a familial condition, it is more commonly a complex trait in which age, sex, genetics, comorbid conditions, and lifestyle factors are key components. Nearly 200 chromosomal loci that influence atrial fibrillation susceptibility have been identified by GWAS, and several atrial fibrillation‐PRS have been devised. Several prospective cohort studies suggest that atrial fibrillation‐PRS are predictive of incident atrial fibrillation (Box) and that addition of atrial fibrillation‐PRS improves predictive ability into models that include age and sex,47 as well as into a model including clinical data such as anthropometrics, blood pressure, smoking status, blood pressure‐lowering medication, diabetes, and history of myocardial infarction and heart failure.48,49,50,51 Atrial fibrillation‐PRS may also be useful for predicting the risk of atrial fibrillation recurrence after ablation procedures.52
Guidelines and screening recommendations
An exponential expansion of technical and analytical capacity has contributed to an increase in genomic research and associations with CVD. However, there is a distinct lack of trial data measuring the impact of genetic test implementation.
Broadly, current guideline recommendations for genetic testing in the clinical setting support its use to confirm causation of a clinical diagnosis for a CVD with established, monogenic inheritance patterns, and to guide cascade genetic and clinical testing in at‐risk family members. Cascade genetic testing refers to a process wherein targeted genetic testing is offered to at‐risk first degree biological relatives of an individual with a germline pathogenic variant; this is typically an iterative process, with further testing of first degree relatives of newly identified probands to reduce incidence, morbidity, health care utilisation, and mortality from certain diseases or conditions.53,54 Individuals should be considered for genetic testing if they have a confirmed or suspected heritable CVD, who are at high a priori risk for a pathogenic variant, and typically following collection of a three‐generation family history and pre‐test genetic counselling.55 A multidisciplinary team with competencies in CVD management and clinical genomics is ideally placed to offer cardiac genetic testing in this setting, considering emerging evidence, recommendations from both medical and clinical genomic societies, and legal or regulatory implications for the patient. Pragmatically, the presenting patient is typically the individual referred for genetic testing; however, the multidisciplinary team should consider proband genetic testing of another affected relative with a more severe phenotype, if available, to increase the chance of identifying a pathogenic variant.
The multidisciplinary team should consider the clinical applicability of genes selected for screening, resisting the inclination to use larger, broader genetic panels due to the more limited clinical application, higher number of variants of unknown significance, and opportunity for incidental secondary genetic findings. The Clinical Genome Resource (ClinGen, https://clinicalgenome.org) provides an up‐to‐date resource for evaluating the contribution of a gene and variant to a disease. Variant classification is a process of considering whether there is sufficient evidence to identify a variant as the cause of disease (ie, pathogenic, likely pathogenic [P/LP]) or not (ie, benign, likely benign). Where there is insufficient or conflicting evidence, an uncertain variant of unknown significance classification will be made. Regular review of variant classifications is necessary, with flow‐on impact to at‐risk relatives who may have used this information in cascade genetic testing.
Pre‐ and post‐test genetic counselling by a trained genetic counsellor is important in ensuring individuals have made a sufficiently informed decision to proceed with testing and have considered potential implications for themselves and their family.56 A summary letter directed towards the relatives, delineating the genetic finding, may be provided to support family communication and the opportunity for at‐risk relatives to consider for cascade clinical and/or genomic testing as appropriate for the condition.
An example of guideline recommendations for genetic testing, and genes currently receiving a “definitive” or “strong” clinical validity classification by ClinGen, are outlined below and presented in the Supporting Information, table 1.
Lipid metabolism
Genetic testing should be considered in patients with total cholesterol of 6.5 mmol/L or greater in the absence of secondary causes, or an LDL‐C of 5–6.5 mmol/L in the context of past medical or family history of premature CAD (Medicare Benefits Schedule [MBS] item 73352).57 Cascade screening of first degree biological family members — involving LDL‐C measurement, targeted variant testing, or both — is also advised (MBS item 73353).
Thoracic aortic disease
Genetic testing for heritable thoracic aortic disease is recommended in individuals with disease diagnosed under 60 years of age or if a positive family history has been identified.58 It should be noted that only 20–30% of causative genes for heritable thoracic aortic disease have been identified. Genetic testing is additionally recommended in patients with suspected Marfan, vascular Ehlers–Danlos, or Loeys–Dietz syndromes to support accurate genetic counselling and screening of at‐risk family members.58 However, no tests are currently Medicare‐rebated and should be completed in the context of specialist clinics.
Inherited cardiomyopathies
Genetic testing is recommended in all patients meeting clinical criteria for hypertrophic cardiomyopathy, dilated cardiomyopathy and arrhythmogenic cardiomyopathy, especially when used to enable cascade screening.59,60,61 Where a patient does not meet the clinical diagnostic criteria, genetic testing is not routinely recommended; however, there may be value for those in whom a monogenic cause is strongly suspected and the testing is performed in a centre with high level expertise, given the high prevalence of variants of unknown significance.56,62 Cascade genetic testing for these conditions in first degree biological family members is recommended if a disease‐causing variant is identified.
Specialists or consultant physicians can request Medicare‐rebated genetic testing for hypertrophic cardiomyopathy, dilated cardiomyopathy or arrhythmogenic cardiomyopathy in patients with a clinical presentation, family history, or laboratory findings consistent with the inherited cardiomyopathy (MBS item 73392). Targeted genetic screening of first degree biological relatives may be requested by specialists or consultant physicians if a P/LP variant is identified (MBS item 73393). Genetic testing may additionally be requested for reproductive partners of the proband to determine reproductive risk (MBS item 73394).
Inherited arrhythmic disorders
Genetic testing is recommended in individuals with known or suspected LQTS based on clinical presentation, family history, and electrocardiogram; genetic testing is additionally recommended in asymptomatic adults with a QTc greater than 500 ms in adults and greater than 480 ms pre‐puberty, in the absence of conditions expected to result in QT prolongation.61 Importantly, genetic testing in LQTS is unique in that results can allow for independent risk prognostication and inform therapeutic decision making.63 Cascade, variant‐specific genetic screening of first degree relatives is recommended if a confident genetic variant is identified in the proband.61
Medicare‐rebated genetic testing for inherited cardiac arrhythmic syndromes or channelopathies may be ordered by specialists or consultant physicians for patients whose clinical presentation or family history indicates a greater than 10% risk of a pathogenic variant (MBS item 73416). If a P/LP variant is identified, specialists or consultant physicians may order variant‐specific testing in first degree biological relatives (MBS item 73417) or reproductive partners (MBS item 73418).
CVD without Mendelian inheritance patterns
Genetic testing in polygenic forms of CVD, including common CVD such as CAD and atrial fibrillation, is still evolving and not currently recommended in a clinical setting.64 Despite strong association of PRS with the relevant disease state, prospective studies collecting evidence on use of these tools are still required for clinical translation. Currently, the role of PRS in patient management remains an active area of research. Close observation of PRS implementation for other non‐cardiovascular conditions, including cancer, will be beneficial.
Medicolegal, insurance and workforce implications
There are several insurance, medicolegal and workforce implications remaining in law and practice which warrant consideration. Globally, consumers report genetic discrimination for insurance coverage as a leading cause of concern when considering testing.65 In Australia, under the Disability Discrimination Act 1992 (Cth),66 genetic information is legally permitted to be incorporated into non‐health insurance policy underwriting, including disability, income protection, life and mortgage insurance.67 The standards and policy framework are self‐regulated by the insurance industry through the Financial Services Council, a members’ association representing more than 100 financial services firms. Since 2019, following a Parliamentary Joint Committee recommendation that genetic discrimination be banned in the life insurance underwriting process,68 the Financial Services Council issued a moratorium on the request for, or use of, genetic information during policy underwriting.69 However, the legal position and ability of insurance providers to use this information during future policy underwriting remains unchanged. Australians have been fortunate for the recent listing of several genetic tests for monogenic cardiac diseases on the MBS, making these accessible at no cost to the patient; clinicians should refer to the MBS Online tool (www.mbsonline.gov.au) for an up‐to‐date list of rebated genetic tests and eligibility criteria. The need for specialised services and genetic counselling to support broad access will greatly increase as more specialists and families wish to pursue this testing.70
Future directions
Incorporation of genomic testing into CVD assessment offers a unique opportunity to further personalise diagnosis and treatment for the individual patient, and, in some cases, target cascade clinical and genomic screening in family members. However, significant gaps remain which impede incorporation of these powerful tools into more routine clinical practice.
Diversity and inclusion in genomic research increase both the groups of individuals benefitting from these novel tools and our scientific understanding of the genetic basis for CVD. Efforts at a regulatory level have sought to increase the diversity and inclusivity of research participants in genetic research. However, validation studies in historically under‐represented ancestry groups frequently demonstrate attenuated or non‐significant results,71,72 and substantial gaps remain in patients from poorly represented ancestry groups, perpetuating health disparities. Numerous studies have shown individuals from diverse ancestry groups have a lower diagnostic yield from genetic testing, due to the higher rate of variants of unknown significance compared with individuals of European ancestry.73
Furthermore, prospective studies and implementation research are necessary to measure the efficacy, health economic, and psychosocial impacts of incorporating genetic testing for CVD diagnosis and management. Networks of academic researchers, primary care and specialty clinicians, clinical geneticists, payers, and health policy experts need to be organised and supported, providing a platform for efficient, prospective clinical implementation studies of emerging tools. One such network, the Partnership for Precision Prevention in CAD, has been formed in Australia, supported by a National Health and Medical Research Council Partnership Grant (GNT2005790). This network has developed a primary care‐based clinical network and launched the first prospective study testing implementation of a PRS‐triaged coronary calcium scan for the diagnosis of subclinical CAD in individuals at low to moderate five‐year CVD risk (Trial No. ACTRN12622000436774).
Conclusion
Substantial advances have been made in the past three decades associating CVD to markers of genetic risk. In some conditions, implementation of genetic testing has improved risk prediction in individual patients, guided treatment decisions, and facilitated cascade screening in at‐risk biological family members, leading to earlier diagnoses and intervention. However, further implementation studies and clinical trials are needed to develop evidence for effective incorporation into the medical practice.
Box – Basic overview of genetic contribution to disease*
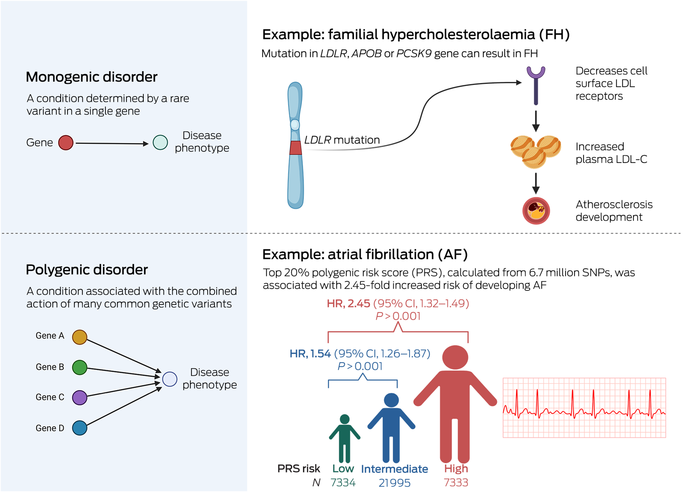
CI = confidence interval; HR = hazard ratio; LDL = low‐density lipoprotein; LDL‐C = low‐density lipoprotein cholesterol; SNP = single‐nucleotide polymorphism. * This can be simplified into monogenic and polygenic disorders, recognising that genetic contribution is a spectrum and many diseases display patterns from both. These patterns are additionally affected by comorbid conditions, lifestyle factors and environmental exposures. Source: Image created with Biorender.com. The data included in the atrial fibrillation example were from Marston et al.50
Provenance: Commissioned; externally peer reviewed.
- Michael P Gray1,2
- Diane Fatkin3
- Jodie Ingles4
- Elizabeth N Robertson1,5
- Gemma A Figtree1,2,6
- 1 University of Sydney, Sydney, NSW
- 2 Kolling Institute, Sydney, NSW
- 3 Victor Chang Cardiac Research Institute, Sydney, NSW
- 4 Agnes Ginges Centre for Molecular Cardiology, Centenary Institute, Sydney, NSW
- 5 Royal Prince Alfred Hospital, Sydney, NSW
- 6 Royal North Shore Hospital, Sydney, NSW
Open access:
Open access publishing facilitated by The University of Sydney, as part of the Wiley ‐ The University of Sydney agreement via the Council of Australian University Librarians.
[Correction added on 8 April 2024, after first online publication: CAUL funding statement has been added.]
Gemma Figtree reports grants from the National Health and Medical Research Council; she is Director and past president of the Australian Cardiovascular Alliance; and is a Board Member of the Heart Foundation. Diane Fatkin is supported by the Heart Foundation and New South Wales (NSW) Health. Elizabeth Robertson is supported by a Translational Research Grant from NSW Health.
Gemma Figtree reports personal fees from CSL; she reports grants from Abbott Diagnostic, Janssen Pharmaceuticals and Sanofi; she is a founding director and Chief Medical Officer of Prokardia; she has a patent for biomarkers and oxidative stress awarded — United States, May 2017 (US9638699B2) — licensed to Northern Sydney Local Health District, a patent Use of P2X7R antagonists in cardiovascular disease (PCT/AU2018/050905) licensed to Prokardia, a patent Methods for treatment and prevention of vascular disease (PCT/AU2015/000548) licensed to the University of Sydney/Northern Sydney Local Health District, and a filed provisional patent application Methods for predicting coronary artery disease (Ref. 2022‐009‐PRO‐0; 2022902660) to the University of Sydney/Northern Sydney Local Health District; she has received personal fees as Executive Committee Member of CPC Clinical Research Jodie Ingles serves as the chair of the ClinGen Hereditary Cardiovascular Disorders Gene Curation Expert Panel.
- 1. World Health Organization. Cardiovascular diseases (CVDs). Geneva: WHO, 2021. https://www.who.int/en/news‐room/fact‐sheets/detail/cardiovascular‐diseases‐(cvds) (viewed Dec 2023).
- 2. Hu P, Dharmayat KI, Stevens CAT, et al. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta‐analysis. Circulation 2020; 141: 1742‐1759.
- 3. Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide prevalence of familial hypercholesterolemia: meta‐analyses of 11 million subjects. J Am Coll Cardiol 2020; 75: 2553‐2566.
- 4. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020; 41: 111‐188.
- 5. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ 1991; 303: 893‐896.
- 6. Elefteriades JA, Sang A, Kuzmik G, Hornick M. Guilt by association: paradigm for detecting a silent killer (thoracic aortic aneurysm). Open Heart 2015; 2: e000169.
- 7. Mészáros I, Mórocz J, Szlávi J, et al. Epidemiology and clinicopathology of aortic dissection. Chest 2000; 117: 1271‐1278.
- 8. Yu HY, Chen YS, Huang SC, et al. Late outcome of patients with aortic dissection: study of a national database. Eur J Cardiothorac Surg 2004; 25: 683‐690.
- 9. Rombouts KB, van Merrienboer TAR, Ket JCF, et al. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest 2022; 52: e13697.
- 10. Renard M, Francis C, Ghosh R, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol 2018; 72: 605‐615.
- 11. Disabella E, Grasso M, Gambarin FI, et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha‐actin 2 (ACTA2). Heart 2011; 97: 321‐326.
- 12. Morisaki H, Akutsu K, Ogino H, et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat 2009; 30: 1406‐1411.
- 13. Wolford BN, Hornsby WE, Guo D, et al. Clinical implications of identifying pathogenic variants in individuals with thoracic aortic dissection. Circ Genom Precis Med 2019; 12: e002476.
- 14. Legrand A, Devriese M, Dupuis‐Girod S, et al. Frequency of de novo variants and parental mosaicism in vascular Ehlers–Danlos syndrome. Genet Med 2019; 21: 1568‐1575.
- 15. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275‐281.
- 16. Lindsay ME, Schepers D, Bolar NA, et al. Loss‐of‐function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 2012; 44: 922‐927.
- 17. Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991; 352: 337‐339.
- 18. Weinsaft JW, Devereux RB, Preiss LR, et al. Aortic dissection in patients with genetically mediated aneurysms: incidence and predictors in the GenTAC Registry. J Am Coll Cardiol 2016; 67: 2744‐2754.
- 19. Wang Y, Starovoytov A, Murad AM, et al. Burden of rare genetic variants in spontaneous coronary artery dissection with high‐risk features. JAMA Cardiol 2022; 7: 1045‐1055.
- 20. Tarr I, Hesselson S, Iismaa SE, et al. Exploring the genetic architecture of spontaneous coronary artery dissection using whole‐genome sequencing. Circ Genom Precis Med 2022; 15: e003527.
- 21. McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol 2021; 18: 22‐36.
- 22. Ingles J, Goldstein J, Thaxton C, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019; 12: e002460.
- 23. Harper AR, Goel A, Grace C, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet 2021; 53: 135‐142.
- 24. Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet 2017; 390: 400‐414.
- 25. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013; 10: 531‐547.
- 26. Jordan E, Peterson L, Ai T, et al. Evidence‐based assessment of genes in dilated cardiomyopathy. Circulation 2021; 144: 7‐19.
- 27. Horvat C, Johnson R, Lam L, et al. A gene‐centric strategy for identifying disease‐causing rare variants in dilated cardiomyopathy. Genet Med 2019; 21: 133‐143.
- 28. Mazzarotto F, Tayal U, Buchan RJ, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 2020; 141: 387‐398.
- 29. Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012; 366: 619‐628.
- 30. Akinrinade O, Koskenvuo JW, Alastalo TP. Prevalence of titin truncating variants in general population. PLoS One 2015; 10: e0145284.
- 31. Fatkin D, Huttner IG, Kovacic JC, et al. Precision medicine in the management of dilated cardiomyopathy: JACC state‐of‐the‐art review. J Am Coll Cardiol 2019; 74: 2921‐2938.
- 32. Fatkin D, Calkins H, Elliott P, et al. Contemporary and future approaches to precision medicine in inherited cardiomyopathies: JACC focus seminar 3/5. J Am Coll Cardiol 2021; 77: 2551‐2572.
- 33. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: executive summary. Heart Rhythm 2019; 16: e373‐e407.
- 34. Peters S, Kumar S, Elliott P, et al. Arrhythmic genotypes in familial dilated cardiomyopathy: implications for genetic testing and clinical management. Heart Lung Circ 2019; 28: 31‐38.
- 35. Kapplinger JD, Landstrom AP, Salisbury BA, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia‐associated mutations from background genetic noise. J Am Coll Cardiol 2011; 57: 2317‐2327.
- 36. James CA, Jongbloed JDH, Hershberger RE, et al. International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the Clinical Genome Resource Framework. Circ Genom Precis Med 2021; 14: e003273.
- 37. Schwartz PJ, Stramba‐Badiale M, Crotti L, et al. Prevalence of the congenital long‐QT syndrome. Circulation 2009; 120: 1761‐1767.
- 38. Ingles J, Semsarian C. Time to rethink the genetic architecture of long QT syndrome. Circulation 2020; 141: 440‐443.
- 39. Newton‐Cheh C, Guo CY, Larson MG, et al. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation 2007; 116: 1128‐1136.
- 40. Schwartz PJ, Ackerman MJ, George AL, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 2013; 62: 169‐180.
- 41. Qureshi SF, Ali A, John P, et al. Mutational analysis of SCN5A gene in long QT syndrome. Meta Gene 2015; 6: 26‐35.
- 42. Zdravkovic S, Wienke A, Pedersen NL, et al. Heritability of death from coronary heart disease: a 36‐year follow‐up of 20 966 Swedish twins. J Intern Med 2002; 252: 247‐254.
- 43. Won HH, Natarajan P, Dobbyn A, et al. Disproportionate contributions of select genomic compartments and cell types to genetic risk for coronary artery disease. PLoS Genet 2015; 11: e1005622.
- 44. Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med 2016; 375: 2349‐2358.
- 45. Inouye M, Abraham G, Nelson CP, et al. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol 2018; 72: 1883‐1893.
- 46. Ball J, Thompson DR, Ski CF, et al. Estimating the current and future prevalence of atrial fibrillation in the Australian adult population. Med J Aust 2015; 202: 32‐35. https://www.mja.com.au/journal/2015/202/1/estimating‐current‐and‐future‐prevalence‐atrial‐fibrillation‐australian‐adult
- 47. Homburger JR, Neben CL, Mishne G, et al. Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome‐wide polygenic scores. Genome Med 2019; 11: 74.
- 48. Okubo Y, Nakano Y, Ochi H, et al. Predicting atrial fibrillation using a combination of genetic risk score and clinical risk factors. Heart Rhythm 2020; 17: 699‐705.
- 49. Kloosterman M, Santema BT, Roselli C, et al. Genetic risk and atrial fibrillation in patients with heart failure. Eur J Heart Fail 2020; 22: 519‐527.
- 50. Marston NA, Garfinkel AC, Kamanu FK, et al. A polygenic risk score predicts atrial fibrillation in cardiovascular disease. Eur Heart J 2023; 44: 221‐231.
- 51. Zhang J, Chen G, Wang C, et al. Associations of risk factor burden and genetic predisposition with the 10‐year risk of atrial fibrillation: observations from a large prospective study of 348,904 participants. BMC Med 2023; 21: 88.
- 52. Al‐Kaisey A, Wong GR, Young P, et al. Polygenic risk scores are associated with atrial electrophysiologic substrate abnormalities and outcomes after atrial fibrillation catheter ablation. Heart Rhythm 2023; 20: 1188‐1194.
- 53. US Centers for Disease Control and Prevention. Cascade testing: finding family members with genetic conditions. Atlanta (GA): CDC, 2022. https://www.cdc.gov/genomics/disease/cascade_testing/cascade_finding.htm (viewed Dec 2023).
- 54. Knowles JW, Rader DJ, Khoury MJ. Cascade screening for familial hypercholesterolemia and the use of genetic testing. JAMA 2017; 318: 381‐382.
- 55. Musunuru K, Hershberger RE, Day SM, et al. Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med 2020; 13: e000067.
- 56. Ingles J, Yeates L, Semsarian C. The emerging role of the cardiac genetic counselor. Heart Rhythm 2011; 8: 1958‐1962.
- 57. Brett T, Arnold‐Reed D. Familial hypercholesterolaemia: a guide for general practice. Aust J Gen Pract 2019; 48: 650‐652.
- 58. Isselbacher EM, Preventza O, Hamilton Black J, et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022; 146: e334‐e482.
- 59. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020; 142: e558‐e631.
- 60. Hershberger RE, Givertz MM, Ho CY, et al. Genetic evaluation of cardiomyopathy — a Heart Failure Society of America practice guideline. J Card Fail 2018; 24: 281‐302.
- 61. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011; 8: 1308‐1339.
- 62. Corrado D, van Tintelen PJ, McKenna WJ, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J 2020; 41: 1414‐1429.
- 63. Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Heart Rhythm 2022; 19: e1‐e60.
- 64. Visseren FLJ, Mach F, Smulders YM, et al. 2021 ESC guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J 2021; 42: 3227‐3337.
- 65. Wauters A, Van Hoyweghen I. Global trends on fears and concerns of genetic discrimination: a systematic literature review. J Hum Genet 2016; 61: 275‐282.
- 66. Australian Government. Disability Discrimination Act 1992. Canberra: Commonwealth of Australia, 1992. https://www.legislation.gov.au/C2004A04426/2018‐04‐12/text (viewed Feb 2024).
- 67. Tiller J, Morris S, Rice T, et al. Genetic discrimination by Australian insurance companies: a survey of consumer experiences. Eur J Hum Genet 2020; 28: 108‐113.
- 68. Parliamentary Joint Committee on Corporations and Financial Services. Inquiry into the Life Insurance Industry. Canberra: Commonwealth of Australia, 2018. https://www.aph.gov.au/Parliamentary_Business/Committees/Joint/Corporations_and_Financial_Services/LifeInsurance/Report (viewed Feb 2024).
- 69. Financial Services Council. FSC Standard No. 11: moratorium on genetic tests in life insurance. Sydney: FSC, 2019. https://www.fsc.org.au/resources‐category/standard/1779‐standard‐11‐moratorium‐on‐genetic‐tests‐in‐life‐insurance/file (viewed Feb 2024).
- 70. Leitch V, Ho E, Ingles J. Genetic counseling — a key first step in genetic testing for people at risk of inherited cardiovascular disease in Australia. Heart, Lung and Circulation 2023; 32: 1239.
- 71. Koyama S, Ito K, Terao C, et al. Population‐specific and trans‐ancestry genome‐wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet 2020; 52: 1169‐1177.
- 72. Coronary Artery Disease (C4D) Genetics Consortium. A genome‐wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 2011; 43: 339‐344.
- 73. Landry LG, Rehm HL. Association of racial/ethnic categories with the ability of genetic tests to detect a cause of cardiomyopathy. JAMA Cardiol 2018; 3: 341‐345.
Summary