





Local guidelines are needed to assist clinicians treating immune thrombocytopenic purpura (ITP) in Australia and New Zealand. Although many excellent summaries have recently been published for audiences elsewhere, we present our accumulated consensus perspectives on the diagnosis and management of ITP, specifically addressing clinically relevant areas where there are limitations to the available evidence1,2,3 (the guideline development process is described in the online Supporting Information, box). We are members of the Thrombosis and Haemostasis Society of Australia and New Zealand (THANZ). This consensus statement has been endorsed by the THANZ Council and ITP Australia. ITP Australia provided patient perspective feedback on our recommendations.
We have used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach to evaluate this evidence and provide recommendations.4 Recommendations have been graded dichotomously as either strong (1) or weak (2), and appended based on the levels of available evidence, rated according to their quality (Box 1).
We followed the internationally accepted guidelines on determining response defined as: “response” if platelet count is ≥ 30 × 109/L and there is at least a twofold increase of the baseline platelet count and absence of bleeding; “complete response” if platelet count is ≥ 100 × 109/L and absence of bleeding.6
Initial investigations
The diagnosis of ITP is one of exclusion, and the extent to which other potential diagnoses need to be excluded varies depending on age, sex and response to treatment. A definitive diagnosis cannot always be made before starting treatment. A response to first line therapy with steroids supports the diagnosis of ITP, whereas absence of response does not exclude ITP but raises the likelihood of alternative causes of thrombocytopenia. Detection of autoantibodies in immune thrombocytopenia is neither sensitive nor specific enough for diagnostic utility.7
The most common alternative diagnoses are myelodysplastic syndrome, familial thrombocytopenia syndromes, hypersplenism, liver disease, and pseudothrombocytopenia.8
Bleeding, family and medication histories and review of historical investigation results are required to gauge bleeding risk and possible hereditary syndromes. A large number of medications and substances can cause thrombocytopenia, such as tonic water (quinine), antibiotics (sulphonamides, chloramphenicol), and alcohol; therefore, a thorough clinical history is critical and reference to comprehensive online resources may be helpful.9,10
Physical examination should focus on assessing for bleeding manifestations as well as splenomegaly and lymphadenopathy. Dysmorphic features are associated with some familial thrombocytopenia syndromes.11
The hallmark laboratory finding of ITP is an isolated thrombocytopenia, and a raised immature platelet fraction can be helpful.12 Microcytic anaemia may be coexistent if thrombocytopenia has contributed to chronic blood loss and subsequent iron deficiency.
Blood film examination is critical in excluding alternative diagnoses, particularly those requiring urgent therapy, such as thrombotic thrombocytopenic purpura (TTP) (Supporting Information, table S1).
Renal and liver function tests should be normal in ITP. New renal dysfunction may raise the possibility of diseases such as a complement‐mediated thrombotic microangiopathy, or TTP. Congenital thrombocytopenia may be suspected if there is a relevant family history, concomitant clinical features, no prior normal platelet count, or lack of response to first line therapy. Consider genetic testing in such patients.
Hepatitis C virus and human immunodeficiency virus (HIV) infections are recognised secondary causes of ITP and without treatment of the underlying virus, the response to therapy can be suboptimal.13
Women of childbearing potential should be investigated for pregnancy. Unlike most autoimmune diseases, ITP tends to flare during pregnancy, and the management of ITP in pregnancy is more constrained due to potential fetal toxicities.14 It is vital that new onset thrombocytopenia occurring later in pregnancy is evaluated expeditiously to exclude pre‐eclampsia and other potentially life‐threatening diseases of pregnancy. Differentiation between ITP and gestational thrombocytopenia can be difficult. An approach to thrombocytopenia in pregnancy has been recently published.15
Detection and eradication of Helicobacter pylori with subsequent remission of ITP has been reported, with highest rates of success in Asian populations.16 In our experience, this has not been successful in most areas of Australia and New Zealand, but may be considered in patients of ethnic backgrounds where supportive data exist (Asia, Middle East, Mediterranean, Latin America), and particularly in those with mild to moderate thrombocytopenia (GRADE 2A).17,18 As eradication is inexpensive and treatment reasonably well tolerated, we also consider testing for H. pylori before performing an irreversible procedure such as splenectomy (GRADE 2C).19
Bone marrow biopsy is not generally required as part of the initial work‐up, unless myelodysplasia needs to be excluded in older patients (aged > 60 years), or in the presence of laboratory abnormalities such as macrocytosis, blood film dysplasia, or after an atypical presentation with slowly deteriorating platelet counts over many years without fluctuation.20 Cytogenetic analysis is recommended (GRADE 1D). We would usually consider performing a bone marrow biopsy before splenectomy (GRADE 2D).
Initial treatment
When to treat
The decision to treat should factor in not only the platelet count but also individual bleeding risk (based on personal history and comorbidities), disease stage (newly diagnosed v persistent or chronic), side effects of treatment, age, concomitant medications, and patient preference.3 We recommend treatment for newly diagnosed ITP when platelet counts are consistently < 20 × 109/L, even in the absence of bleeding (GRADE 1C).21,22 If the patient has no or only mild bleeding and platelets > 20 × 109/L, then a watch‐and‐wait strategy is usually appropriate (GRADE 2C).23
First line treatment
Steroids
Steroids are the standard first line treatment, usually prednisone or dexamethasone. Several regimens are used, but if choosing prednisone, we recommend a starting dose of 1 mg/kg/day for the first 2 weeks, followed by a tapering regimen over 6 weeks (GRADE 1C).24 Consider initially capping the dose to 75–80 mg once daily, even for patients weighing > 80kg (GRADE 2D).1
An alternative regimen is dexamethasone 40 mg or 0.6 mg/kg orally once daily for 4 days, every 14–28 days for one to six cycles (GRADE 1C).3,25 Some investigators report higher remission rates with pulsed dexamethasone as opposed to standard‐dose prednisone, with fewer adverse effects.26,27 The dexamethasone dose can be attenuated to 20 mg for older patients (GRADE 2D).
Patients requiring longer term steroid therapy (steroid‐dependent after more than 8–10 weeks) or repeated courses of steroid therapy should be referred to a tertiary centre with experience in ITP (GRADE 2D).
Our panel agrees there is clinical equipoise between prednisone versus dexamethasone, with prednisone favoured in older patients less likely to tolerate the neuropsychiatric side effects of dexamethasone, and dexamethasone favoured in those seeking a more rapid response with shorter overall duration of steroid exposure.
Intravenous immunoglobulin (IVIg)
IVIg can be used periprocedurally as on‐demand or as first line therapy in combination with steroids. The 5% and 10% formulations appear to have similar efficacy (response rates about 75%).28 Dosing options include 0.4 g/kg daily for 3–5 days or 1 g/kg for 1–2 days, with the latter option being associated with a faster response.1,29 Therapy with prednisone or dexamethasone can be combined with IVIg, or intravenous methylprednisolone can be substituted for the oral steroid, if there is a need for a more rapid response (GRADE 1C).30 In Australia, IVIg availability is facilitated through the BloodSTAR program and in New Zealand via New Zealand Blood Service. Criteria permitting access to IVIg generally require thrombocytopenia < 30 × 109/L, the presence or perceived risk of bleeding, poor response to other therapies (steroids in newly diagnosed ITP or splenectomy in chronic ITP), and special clinical circumstances (pregnancy, periprocedural).31 In New Zealand, IVIg is available as first line therapy at the discretion of the haematologist. Subcutaneous immunoglobulin and anti‐D are not available in Australia and New Zealand for ITP.
Second line treatments
There is no consensus on which second line treatment for ITP should be attempted first. There is also no reliable predictor of response to second line treatments. Patients should switch to a second line treatment when the first line treatment has not obtained a haemostatic response. The risk of bleeding and mortality increases with platelet counts < 20 × 109/L; hence, treatment is often aimed at achieving a platelet count > 20 × 109/L and avoidance of severe bleeding.24,32
Inadequate haemostatic response with > 5 mg/day of prednisone, three to four cycles of high dose dexamethasone, or with one or more courses of IVIg represent failure of first line treatment.1,3
Patient preferences, age, lifestyle, comorbidities, and drug availability are important when considering when to start a second line treatment and which treatment modality to adopt (GRADE 1D).
Splenectomy, rituximab and thrombopoietin receptor agonists (TPO‐RAs) have the most robust evidence in terms of efficacy and safety and hence it is recommended that these three options be discussed with patients as suitable second line treatments (GRADE 1C)33 (Box 2). Some patients are reluctant to undergo splenectomy if there is a non‐surgical alternative.34,35 Public reimbursement for TPO‐RAs in Australia and New Zealand is limited to later lines of therapy unless there are medical contraindications to splenectomy.
Given that up to one‐third of patients achieve a satisfactory haemostatic response (either spontaneously or with treatment), we agree with most other international guidelines on ITP that splenectomy be delayed for at least 12 months (GRADE 1C).1,36 Therefore, for patients with ITP with a disease duration of less than 12 months, TPO‐RAs and rituximab should be pursued as first choice second line treatment. If TPO‐RAs and rituximab are not accessible or have failed, other options need to be explored, as described below.
Splenectomy
Efficacy
Splenectomy is associated with the greatest likelihood of durable remission, with a long term response rate of 60–70%.37,38 Splenic patterns of uptake on indium‐labelled autologous platelet scanning have been reported to be predictive of splenectomy response, but this radioisotope is difficult to obtain in Australasia.39,40,41 Higher relapse rates to splenectomy have been reported in older patients (aged > 65 years).42
Adverse events
Infections and thromboembolism are the main complications associated with splenectomy, both acutely and longer term.37,42 Patients aged over 65 years are more susceptible to these complications.42,43 Laparoscopic splenectomy is associated with shorter hospitalisation stay and reduced perioperative bleeding and patient discomfort compared with laparotomy.44,45
Vaccinations against encapsulated bacteria Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae should be administered before splenectomy and rituximab when possible (GRADE 1C).46 Postoperative thromboprophylaxis and antibiotic prophylaxis should be administered as per local and national guidelines for splenectomy, but patients with ITP are at a higher risk of thrombosis. For patients living in Victoria, Queensland and Tasmania, patient registration with Spleen Australia (http://spleen.org.au) can assist in optimising follow‐up and surveillance.
Patient selection
Splenectomy should be considered in patients aged less than 65 years, with disease duration greater than 12 months, and for whom this option impacts least on their lifestyle (GRADE 2D).1,42 Patients without a history of thrombosis or infections are favourable candidates for splenectomy (GRADE 2D).
Rituximab
Efficacy
Rituximab is an antibody directed against CD20, leading to B lymphocyte depletion. Early (1–2 weeks) and late (8–12 weeks) responses have been observed with rituximab.47 While 50–70% of patients achieve an initial response to rituximab monotherapy, the long term response rate of 20–25% is less and lower than splenectomy.48 Response rates are reported to be higher in females, in patients with shorter disease duration (< 1–2 years), in younger patients (aged < 40 years), and when combined with dexamethasone.49,50
Administration and dosing
Rituximab dosing regimens administered in ITP include 375 mg/m2/week for 4 weeks, 1 g rituximab on day 1 and day 15, and 100 mg/week for 4 weeks.51,52 Time to response has been reported to be slower in lower dose strategies, but there is no advantage in response rate with standard or higher dose regimens.53 While in Australia rituximab is not reimbursed for ITP by the Pharmaceutical Benefits Scheme (PBS), the advent of rituximab biosimilars has reduced the cost of rituximab, and we anticipate this will improve access through local institutions. In New Zealand, rituximab is available on the Pharmaceutical Management Agency (PHARMAC) before splenectomy or in refractory cases. Readministration of rituximab can be considered in patients who have obtained an initial response of more than 12 months (GRADE 2C).54
Adverse events
Rituximab is generally well tolerated. Infusion reactions are more likely to occur with standard dose or 1 g rituximab infusions, and successful B‐cell depletion is associated with an increased risk of infections. A serious but very rare complication of rituximab is progressive multifocal leukoencephalopathy. Hepatitis B carrier status should be reviewed before treatment commencement due to the risk of reactivation (GRADE 1C).55 Vaccine responses can be suppressed by rituximab for up to 6 months; therefore, potential candidates for subsequent splenectomy in rituximab failure should be offered vaccinations before commencing rituximab therapy (GRADE 1D).56
Patient selection
Rituximab should be considered in patients who have expressed a strong preference to avoid surgery. We recommend administering rituximab with high dose dexamethasone (up to three cycles) (GRADE 1C).49 Rituximab is favoured for patients without a concomitant immunodeficiency and for those at risk of thrombosis (GRADE 1D). Rituximab should be considered in younger, female patients with short disease duration (< 1–2 years) (GRADE 2C).49
TPO‐RAs: eltrombopag and romiplostim
Efficacy
Eltrombopag and romiplostim have been validated in randomised placebo‐controlled trials in patients with persistent and chronic ITP, showing response rates of 60–90% as early as 2–3 weeks.57,58 However, durable responses with persisting robust platelet counts is lower and in the order of 40–60%.59,60 Nevertheless, TPO‐RAs significantly reduce the incidence of severe bleeding and the need for rescue therapy, and improve health‐related quality of life measures.61,62
Administration and dosing
Eltrombopag is given once daily orally while romiplostim is dosed as a weekly subcutaneous injection. Eltrombopag dosing starts at 50 mg daily (25 mg daily in East Asian people) and can be increased up to a maximum of 75 mg daily.63 Eltrombopag must be given on an empty stomach; in particular, it should be taken 4 hours after or 2 hours before products or food containing cations (calcium, dairy products, iron supplements).
Romiplostim dosing starts at 1 µg/kg/week, and can be increased up to a maximum of 10 µg/kg/week until a response is achieved. In cases where a rapid response is needed, we recommend starting with the contents of one small vial (250–375 µg), which is often approximately 3–4 µg/kg (GRADE 1D).
Approval for continuing PBS reimbursement after 24 weeks for TPO‐RAs requires demonstration of a platelet‐count response. In Australia, this response is defined as four separate platelet counts i) ≥ 50 × 109/L or ii) > 30 × 109/L and double baseline platelet count; in New Zealand, response is defined as platelet counts ≥ 50 × 109/L.
Unfortunately, these response definitions fail to recognise a clinically observed response where platelet counts improve, patients experience less bleeding, but the platelet counts may not meet strict response criteria. It is helpful to document the lowest baseline platelet count before TPO‐RAs commencement to meet these criteria subsequently (GRADE 2D). In addition, we recommend repeat platelet count testing frequently to establish the required evidence of response for PBS reimbursement rather than abandoning therapy, as platelet counts can fluctuate in some cases (GRADE 2D). Without resorting to rescue therapies, adjunctive strategies may also be helpful to support platelet counts to help meet PBS criteria for reimbursement (Box 3) (GRADE 2D).
Treatment‐free response and discontinuation
About 10–30% of patients taking TPO‐RAs are able to discontinue and maintain a treatment‐free response.72 Predictors for treatment‐free response are not established; however, TPO‐RAs discontinuation may be considered in patients maintaining platelet responses > 50 × 109/L for more than 6–12 months, absence of previous major bleeding, and/or requiring only low doses of TPO‐RAs (GRADE 2D). Given the risk of rebound thrombocytopenia, TPO‐RAs discontinuation should not occur abruptly, but it should be done with a slow taper. We recommend that discontinuation be delayed if there is a history of significant platelet count fluctuation, variable adherence to therapy, past major bleeding, or sudden relapse (GRADE 1D). Tapering to cessation can be commenced sooner if platelet counts are persistently above 200 × 109/L (GRADE 2D). In contrast, about 30% of patients discontinue TPO‐RAs because of a lack of response.73 Switching from one TPO‐RA to another has been shown to be effective for some patients and is permissible for PBS reimbursement with written application.61
Adverse events
Reports of increased bone marrow fibrosis by TPO‐RAs have raised concerns of myelofibrosis risk, but clinically significant myelofibrosis is rare.61 An increased risk of venous and arterial thrombosis has been observed in patients with ITP treated with TPO‐RAs compared with patients with ITP without TPO‐RAs exposure. These observations are largely based on registry and retrospective studies, and patients with a history of thrombosis should be informed of this risk (GRADE 2C).59,74 Adverse events more commonly observed with eltrombopag include transaminitis, and liver function monitoring is recommended (GRADE 1B).61
Patient selection
In Australia, TPO‐RAs are approved for treatment of chronic ITP following splenectomy and inadequate response to IVIg, or in patients for whom splenectomy is contraindicated. In New Zealand, TPO‐RAs are approved as fourth line therapy after splenectomy or as third line therapy if splenectomy is contraindicated.
We recommend romiplostim in patients with gastrointestinal diseases, abnormal liver function, or who are unable to adhere to prescribed dietary restrictions (GRADE 1D). We recommend eltrombopag in patients who have a needle phobia and in those who prefer the simplicity of once daily dosing (GRADE 1D). Paradoxically, as eltrombopag can be difficult to administer effectively in an aged care environment due to the uncertainty of dose timing in relation to meal service, we recommend romiplostim in this setting (GRADE 2D).
Other therapies
For patients for whom rituximab, TPO‐RAs or splenectomy are not accessible or have an unfavourable risk–benefit profile, there are a number of alternative medications that can be considered (Box 3).
Mycophenolate mofetil and dapsone are preferred in this setting, with a quicker onset of action compared with the other medications (GRADE 1C).75,76 While these drugs are generally more accessible and affordable, evidence regarding efficacy is less robust. Their onset of action can be more prolonged than that of rituximab, splenectomy or TPO‐RAs. In view of this, steroids are often administered concurrently with these medications while awaiting response, resulting in additional toxicity. Prolonged treatment may also be required, and alternative strategies should be quickly considered if toxicity is encountered.
While there is currently no Australian or New Zealand supplier of danazol, in Australia the drug can be accessed by the Special Access Scheme, which is approved by the Therapeutic Goods Administration.
For further discussion on less commonly used alternatives, the reader may refer to Cuker and Neunert.68
Beyond second line therapies
Clinical trial enrolment of eligible patients with ITP who have not responded to currently available therapies is strongly recommended where available in limited sites around Australia and New Zealand (GRADE 1D).
Imaging could be performed in patients who initially achieved a response after splenectomy but subsequently relapsed to exclude the development of an accessory spleen.77
Non‐rescue low dose steroids, such as prednisolone ≤ 5 mg/day or 10 mg once a week, can be considered to improve the response to many second line therapies, including TPO‐RAs (GRADE 2D). Combination therapy can also have a synergistic effect (eg, TPO‐RAs plus an immunosuppressant such as mycophenolate mofetil or azathioprine) (GRADE 2D).78
Supportive care in ITP
The supportive care of patients with ITP includes management of acute bleeding, avoidance of long term side effects of therapies (particularly steroids), and identification of fatigue. Adjunctive therapies that may be helpful in acute bleeding include tranexamic acid (avoid if haematuria) and proton pump inhibitors in major gastrointestinal bleeding.1
Avoidance of long term high dose steroid use in patients with ITP is imperative (GRADE 1B).79 Patients taking steroids should be monitored for known side effects, including hyperglycaemia, mood or sleep disturbance, osteopenia, and infection. Patients taking an equivalent of prednisolone 20 mg daily for more than 2 weeks are at increased risk of infection, and the Therapeutic Guidelines provide further recommendations for such patients.80
Bone mineral density assessments should be considered for patients who have received prolonged steroids and are at risk of osteopenia, such as post‐menopausal women, and they should be proactively managed with calcium and vitamin D supplements. Currently, PBS reimbursement exists for intravenous zoledronic acid 5 mg annually with corticosteroid‐induced osteopenia (GRADE 2D).
Fatigue appears common in patients with ITP, but its optimal management has not been ascertained.81 Referral to counselling and ITP‐specific patient support networks may be helpful.
Splenectomised patients should be reviewed for their risk of infection and thrombosis, adherence to local guidelines on long term antibiotic use, immunisation status, and modifiable vascular risk factors (GRADE 1D).
Special situations
Treatment of emergency bleeding in ITP
Life‐threatening bleeding, such as intracranial haemorrhage, has been reported in 0.1–0.4% of children and 1.4% of adults.82 Severe bleeding is reported in 9.5% of adults.82,83 When considering the treatments and outcomes for patients with acute life‐threatening bleeding in ITP, the wide range of options underpins the lack of evidence.84 We therefore recommend a number of measures introduced simultaneously rather than sequentially (GRADE 1D).85,86,87
In life‐threatening bleeding, we recommend platelet transfusions to achieve haemostasis, along with IVIg (1–2 g/kg), and steroids (methylprednisolone up to 1000 mg intravenous daily for 1–5 days or high dose dexamethasone 40 mg daily intravenous or orally for 4 days) (Grade 1D).22,88,89
Vinka alkaloids can be considered for rare cases of refractory or multiply relapsed disease and life‐threatening bleeding (GRADE 2D). Some authors have experience with using vincristine 1–2 mg intravenously (over 4–6 hours) weekly for two to four doses, and treatment effect can be seen in less than 48 hours.90
Supportive red cell transfusions, antifibrinolytic therapy with tranexamic acid (up to 1000 mg intravenous three times a day) and other blood products may be useful.3,22 Local measures such as endoscopic cautery need to be considered in gastrointestinal bleeding and epistaxis (GRADE 1D).
Splenectomy for emergency bleeding is difficult given the dangers of unplanned surgery.91
Platelet thresholds for planned interventions in ITP
Stable patients with ITP may require a rapid increment in their platelet counts for scenarios such as emergency or elective surgery or imminent childbirth. Although there is little direct evidence in ITP, we support a risk–benefit approach to platelet targets dependent on the intervention proposed (Supporting Information, table S2).
Therapeutic interventions to raise platelet counts before surgery or procedures
For emergency procedures (within hours), IVIg (1 g/kg) with intravenous methylprednisolone (500–1000 mg) should be given immediately (GRADE 1B),86,87 and platelet transfusion (at induction of anaesthesia, and subsequently intra‐ and/or postoperatively depending on bleeding) should be given as close to the time of the procedure as possible or on induction of anaesthesia, with expected platelet survival of 1–4 hours (GRADE 1C).92 Do not delay any procedure to confirm a platelet increment, as very little would be expected (GRADE 1D). Repeat doses of IVIg may be needed if postoperative bleeding risk remains high.
For elective procedures (days to one week), options include IVIg (GRADE 1B),93 steroids (GRADE 1B),87 or TPO‐RA (romiplostim 500 µg subcutaneous weekly for two doses; commencing 10 days before surgery) (GRADE 1D).
Pregnancy
In the first and second trimesters, the indication for treatment is a platelet count < 20 × 109/L. For vaginal or Caesarean delivery, a platelet count ≥ 50 × 109/L is generally adequate.94
For women with platelet counts < 20 × 109/L, prednisone 50 mg daily could be considered (GRADE 2D). If IVIg is used before delivery or for life‐threatening haemorrhage, the recommended dose is 1–2 g/kg as a single or divided dose (GRADE 1D). Patients may respond to the combination of steroids and IVIg if they do not respond to monotherapy. It is sometimes useful to rehearse ITP treatment several weeks before term, in order to plan for a neuraxial anaesthesia, where a platelet target ≥ 70 × 109/L is reasonable (GRADE 2D).
We suggest referring to the Haematology in Obstetrics and Women’s Health (HOW Collaborative) guidelines on managing thrombocytopenia in pregnancy for assistance on pregnant patients who fail first line therapies.95
Arterial disease and severe thrombocytopenia in ITP
Older patients are being increasingly recognised and diagnosed with ITP.96 The increased prevalence of vascular disease in older patients becomes difficult to manage in more severe thrombocytopenia with ITP.97
Second line immunosuppressive therapies may be more attractive, as splenectomy would be less likely to be safe in patients with vascular comorbidities, in addition to the inherent increased risk of thrombosis following splenectomy.97 After balancing the risk of vascular disease against the risk of bleeding, the clinician is advised to target the greater problem; if they are both unacceptable, we advise increasing ITP therapy to mitigate the risk of bleeding from treating the vascular disease (GRADE 2D).2
It is generally safe to administer antiplatelet therapy if platelet counts are ≥ 30 × 109/L, and dual antiplatelet therapy if platelet counts are ≥ 50 × 109/L (GRADE 1D). Bare metal stents may reduce the duration of antiplatelet therapy required and may be preferred in patients with unstable or refractory thrombocytopenia (GRADE 2D). Anticoagulation for atrial fibrillation can be considered when the likely benefits outweigh the risk of bleeding, and it is usually safe with platelet counts ≥ 50 × 109/L, but may be individualised in patients without a history of thrombocytopenic bleeding down to 30 × 109/L (GRADE 1D).
Platelet transfusions have no role in routinely supporting platelet counts for antiplatelet therapies or anticoagulation in patients with ITP (GRADE 1D).
Venous thromboembolic disease and severe thrombocytopenia in ITP
The diagnosis and treatment of venous thromboembolic disease (VTE) can be complicated by ITP. Severe thrombocytopenia can be misunderstood as a negative risk factor for thrombosis, delaying the time to diagnosis. Paradoxically, both ITP and its treatments may increase the risk for VTE.98,99,100
It is much easier to administer prophylaxis against deep vein thrombosis than to treat established thrombosis in severe thrombocytopenia. Prophylactic doses of low molecular weight heparin are generally safe to administer with platelet counts ≥ 30 × 109/L (GRADE 1D).
Therapeutic anticoagulation is generally safe to administer for VTE management with platelet counts ≥ 30 × 109/L, and reduced intensity (half dose) anticoagulation is probably safe for platelet counts 20–30 × 109/L (GRADE 1D).101,102,103 The duration of anticoagulation and selection of anticoagulant are mostly unaffected by ITP, although patients with unstable platelet counts or a history of recent bleeding may be safer on anticoagulants with reversibility such as vitamin K antagonists (warfarin) and dabigatran, which is not reimbursed by the PBS in Australia, but it is funded by PHARMAC in New Zealand for VTE (GRADE 2D). There is increasing familiarity with direct oral anticoagulants, but these should be used with caution in patients with labile platelet counts.
ITP therapy can be titrated to raise the platelet count for safer anticoagulation, but this should be balanced against the risk of provoking VTE (GRADE 2D). Prothrombotic ITP therapies (eg, TPO‐RA) can usually be continued while remaining on indefinite anticoagulation (GRADE 2D). After excluding other causes of VTE (eg, antiphospholipid syndrome), strong consideration should be given to changing ITP therapies if the thromboembolic event was life‐threatening (GRADE 1D).
Conclusions
Most recommendations are made with low Levels of Evidence (Supporting Information, table S4). Paradoxically, this is not a weakness of these guidelines, but instead the quintessential raison d’être for these guidelines targeted at the Australasian audience. The absence of high quality evidence for basic clinical dilemmas in ITP underlines the need for updated guidelines relevant to the local treatment context, as well as ongoing collaborative scientific and clinical research in ITP (Supporting Information, table S3).
We stand on the precipice of a new age in the treatment of ITP, with novel therapeutics targeting Fc neonatal receptors, Bruton tyrosine kinase (BTK) and spleen tyrosine kinase (SYK) signalling, and complement inhibition at varying stages of promising research maturity.104,105,106,107 Anticipating the best way to incorporate these new modalities into our already crowded but flawed treatment armamentarium remains a challenge for us in the future.
Updates of these guidelines are anticipated as major milestone advances in therapy become available in the Australasian market in the coming years.
Box 1 – Grading system employed to append recommendations*
|
Strength of recommendation |
|
||||||||||||||
|
|
|||||||||||||||
|
1 |
Strong |
||||||||||||||
|
2 |
Weak |
||||||||||||||
|
Levels of Evidence |
|
||||||||||||||
|
A |
High quality meta‐analysis |
||||||||||||||
|
B |
Robust phase 3 studies |
||||||||||||||
|
C |
Well designed phase 2 studies, or good quality case series |
||||||||||||||
|
D |
Expert panel consensus |
||||||||||||||
|
|
|||||||||||||||
|
*Strength of recommendation and grading of evidence as reviewed by the expert panel from 1 January 1996 to 1 February 2021.4,5 Statements graded (1A) are strong consensus recommendations supported by high quality meta‐analyses, while statements graded (2D) are weak opinions unsupported by any evidence but agreed upon by all authors. |
|||||||||||||||
Box 2 – Management of immune thrombocytopenic purpura with second line therapies and beyond
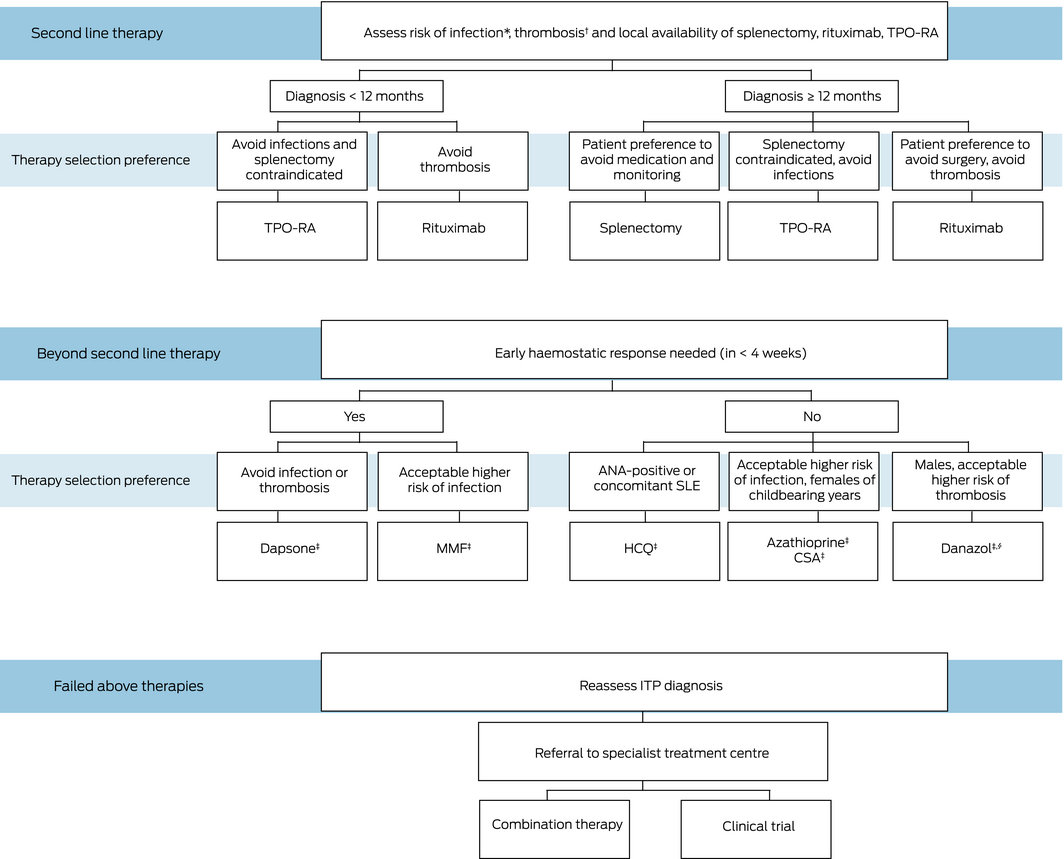
CSA = ciclosporin; HCQ = hydroxychloroquine; MMF = mycophenolate mofetil; SLE = systemic lupus erythematosus; TPO‐RA = thrombopoietin receptor agonists. * Patient with acute or chronic infections or a past history of life threatening infection. † Patient with a past history of thrombosis. ‡ Steroids are often administered concurrently with these medications while awaiting response. § Access issues as described in text above.
Box 3 – Other long term therapy options for adult patients with immune thrombocytopenic purpura
|
Medication |
Funding |
Patient selection |
Recommended dose and treatment strategy |
Response rate |
Time to response |
Toxicities |
|||||||||
|
|
|||||||||||||||
|
Azathioprine64 |
|
|
40–60% |
3–4 months |
|
||||||||||
|
Mycophenolate mofetil66 |
|
|
|
50–60% |
50% of patients respond by 4 weeks |
|
|||||||||
|
Hydroxychloroquine67 |
|
|
|
60% |
2–3 months |
|
|||||||||
|
Danazol68 |
|
|
|
40–50% |
3–6 months |
|
|||||||||
|
Dapsone69 |
|
|
|
50% |
3 weeks |
|
|||||||||
|
Ciclosporin70 |
|
|
|
40–60% |
1–3 months |
|
|||||||||
|
|
|||||||||||||||
|
ANA = antinuclear antibodies; FBC = full blood count; G6PD = glucose‐6‐phosphate dehydrogenase; LDH = lactate dehydrogenase; PBS = Pharmaceutical Benefits Scheme (Australia); PHARMAC = Pharmaceutical Management Agency (New Zealand); PSA = prostate‐specific antigen; SAS = Special Access Scheme; TPMT = thiopurine methyltransferase; TPO‐RAs = thrombopoietin receptor agonists. |
|||||||||||||||
Provenance: Not commissioned; externally peer reviewed.
- Philip YI Choi1,2
- Eileen Merriman3
- Ashwini Bennett4,5
- Anoop K Enjeti6,7
- Chee Wee Tan8,9
- Isaac Goncalves10,11
- Danny Hsu12,13
- Robert Bird14
- 1 Canberra Hospital, Canberra, ACT
- 2 Australian National University, Canberra, ACT
- 3 Waitematā District Health Board, Auckland, New Zealand
- 4 Monash Medical Centre, Melbourne, VIC
- 5 Monash University, Melbourne, VIC
- 6 Calvary Mater Hospital, Newcastle, NSW
- 7 University of Newcastle, Newcastle, NSW
- 8 Royal Adelaide Hospital, Adelaide, SA
- 9 SA Pathology, Adelaide, SA
- 10 Peter MacCallum Cancer Centre, Melbourne, VIC
- 11 Royal Melbourne Hospital, Melbourne, VIC
- 12 South Western Sydney Local Health District, Sydney, NSW
- 13 University of New South Wales, Sydney, NSW
- 14 Princess Alexandra Hospital, Brisbane, QLD
Our work to write these guidelines is not funded. THANZ provided support for online open access publication costs.
Philip Choi received speaking fees for lectures and presentations for Amgen Australia.
- 1. Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv 2019; 3: 3780–3817.
- 2. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv 2019; 3: 3829–3866.
- 3. Matzdorff A, Meyer O, Ostermann H, et al. Immune thrombocytopenia — current diagnostics and therapy: recommendations of a Joint Working Group of DGHO, OGHO, SGH, GPOH, and DGTI. Oncol Res Treat 2018; 41 (Suppl): 1–30.
- 4. Balshem H, Helfand M, Schünemann HJ, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol 2011; 64: 401–406.
- 5. Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008; 336: 924–926.
- 6. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113: 2386–2393.
- 7. Brighton TA, Evans S, Castaldi PA, et al. Prospective evaluation of the clinical usefulness of an antigen‐specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood 1996; 88: 194–201.
- 8. Arnold DM, Nazy I, Clare R, et al. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: lessons from the McMaster ITP Registry. Blood Adv 2017; 1: 2414–2420.
- 9. Chong BH, Choi PY, Khachigian L, Perdomo J. Drug‐induced immune thrombocytopenia. Hematol Oncol Clin North Am 2013; 27: 521–540.
- 10. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug‐induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009; 7: 911–918.
- 11. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004; 103: 390–398.
- 12. Ferreira FLB, Colella MP, Medina SS, et al. Evaluation of the immature platelet fraction contribute to the differential diagnosis of hereditary, immune and other acquired thrombocytopenias. Sci Rep 2017; 7: 3355.
- 13. Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113: 6511–6521.
- 14. Gernsheimer T, James AH, Stasi R. How I treat thrombocytopenia in pregnancy. Blood 2013; 121: 38–47.
- 15. Eslick R, Cutts B, Merriman E, et al. HOW Collaborative position paper on the management of thrombocytopenia in pregnancy. Aust N Z J Obstet Gynaecol 2021; 61: 195–204.
- 16. Lee A, Hong J, Chung H, et al. Helicobacter pylori eradication affects platelet count recovery in immune thrombocytopenia. Sci Rep 2020; 10: 9370.
- 17. Kim BJ, Kim HS, Jang HJ, Kim JH. Helicobacter pylori eradication in idiopathic thrombocytopenic purpura: a meta‐analysis of randomized trials. Gastroenterol Res Pract 2018; 2018: 6090878.
- 18. Pezeshki SMS, Saki N, Ghandali MV, et al. Effect of Helicobacter pylori eradication on patients with ITP: a meta‐analysis of studies conducted in the Middle East. Blood Res 2021; 56: 38–43.
- 19. Sivapathasingam V, Harvey MP, Wilson RB. Helicobacter pylori eradication: a novel therapeutic option in chronic immune thrombocytopenic purpura. Med J Aust 2008; 189: 367–370. https://www.mja.com.au/journal/2008/189/7/helicobacter‐pylori‐eradication‐novel‐therapeutic‐option‐chronic‐immune
- 20. Waisbren JB, Dinner SN, Helenowski I, et al. Disease characteristics and prognosis of myelodysplastic syndrome presenting with isolated thrombocytopenia. Blood 2015; 126: 3477–3477.
- 21. Sirotich E, Guyatt G, Gabe C, et al. Definition of a critical bleed in patients with immune thrombocytopenia: Communication from the ISTH SSC Subcommittee on Platelet Immunology. J Thromb Haemost 2021; 19: 2082–2088.
- 22. Arnold DM. Bleeding complications in immune thrombocytopenia. Hematology Am Soc Hematol Educ Program 2015; 2015: 237–242.
- 23. Cines DB, Bussel JB. How I treat idiopathic thrombocytopenic purpura (ITP). Blood 2005; 106: 2244–2251.
- 24. Piel‐Julian ML, Mahévas M, Germain J, et al; CARMEN Investigators Group. Risk factors for bleeding, including platelet count threshold, in newly diagnosed immune thrombocytopenia adults. J Thromb Haemost 2018; 16: 1830–1842.
- 25. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood 2017; 129: 2829–2835.
- 26. Wei Y, Ji XB, Wang YW, et al. High‐dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016; 127: 296–302; quiz 370.
- 27. Matschke J, Muller‐Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment‐naive adult patients with immune thrombocytopenia: EIS 2002 Study. Acta Haematologica 2016; 136: 101–107.
- 28. Robak T, Mainau C, Pyringer B, et al. Efficacy and safety of a new intravenous immunoglobulin 10% formulation (octagam 10%) in patients with immune thrombocytopenia. Hematology 2010; 15: 351–359.
- 29. Kovaleva L, Apte S, Damodar S, et al. Safety and efficacy of a 10% intravenous immunoglobulin preparation in patients with immune thrombocytopenic purpura: results of two international, multicenter studies. Immunotherapy 2016; 8: 1371–1381.
- 30. Godeau B, Chevret S, Varet B, et al; French ATIP Study Group. Intravenous immunoglobulin or high‐dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: a randomised, multicentre trial. Lancet 2002; 359: 23–29.
- 31. IG Governance; BloodSTAR. Criteria for clinical use of immunoglobulin in Australia. Immune thrombocytopenic purpura (ITP) — adult; version 3.1 [website]. National Blood Authority; 2018. https://www.criteria.blood.gov.au/MedicalCondition/View/2571 (viewed Dec 2020).
- 32. McMillan R, Durette C. Long‐term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104: 956–960.
- 33. Lal LS, Said Q, Andrade K, Cuker A. Second‐line treatments and outcomes for immune thrombocytopenia: a retrospective study with electronic health records. Res Pract Thromb Haemost 2020; 4: 1131–1140.
- 34. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016; 91: E267–E272.
- 35. Choi PY, Gordon JE, Harvey M, Chong BH. Presentation and outcome of idiopathic thrombocytopenic purpura in a single Australian centre. Intern Med J 2012; 42: 841–845.
- 36. Sailer T, Lechner K, Panzer S, et al. The course of severe autoimmune thrombocytopenia in patients not undergoing splenectomy. Haematologica 2006; 91: 1041–1045.
- 37. Vecchio R, Marchese S, Intagliata E, et al. Long‐term results after splenectomy in adult idiopathic thrombocytopenic purpura: comparison between open and laparoscopic procedures. J Laparoendosc Adv Surg Tech A 2013; 23: 192–198.
- 38. Vianelli N, Palandri F, Polverelli N, et al. Splenectomy as a curative treatment for immune thrombocytopenia: a retrospective analysis of 233 patients with a minimum follow up of 10 years. Haematologica 2013; 98: 875–880.
- 39. Navez J, Hubert C, Gigot JF, et al. Does the site of platelet sequestration predict the response to splenectomy in adult patients with immune thrombocytopenic purpura? Platelets 2015; 26: 573–576.
- 40. Roca M, Muniz‐Diaz E, Mora J, et al. The scintigraphic index spleen/liver at 30 minutes predicts the success of splenectomy in persistent and chronic primary immune thrombocytopenia. Am J Hematol 2011; 86: 909–913.
- 41. Sarpatwari A, Provan D, Erqou S, et al. Autologous 111 in‐labelled platelet sequestration studies in patients with primary immune thrombocytopenia (ITP) prior to splenectomy: a report from the United Kingdom ITP Registry. Br J Haematol 2010; 151: 477–487.
- 42. Gonzalez‐Porras JR, Escalante F, Pardal E, et al. Safety and efficacy of splenectomy in over 65‐yrs‐old patients with immune thrombocytopenia. Eur J Haematol 2013; 91: 236–241.
- 43. Massarweh NN, Legner VJ, Symons RG, et al. Impact of advancing age on abdominal surgical outcomes. Arch Surg 2009; 144: 1108–1114.
- 44. Tada K, Ohta M, Saga K, et al. Long‐term outcomes of laparoscopic versus open splenectomy for immune thrombocytopenia. Surg Today 2018; 48: 180–185.
- 45. Qu Y, Xu J, Jiao C, et al. Long‐term outcomes of laparoscopic splenectomy versus open splenectomy for idiopathic thrombocytopenic purpura. Int Surg 2014; 99: 286–290.
- 46. Bonanni P, Grazzini M, Niccolai G, et al. Recommended vaccinations for asplenic and hyposplenic adult patients. Hum Vaccin Immunother 2017; 13: 359–368.
- 47. Stasi R, Stipa E, Forte V, et al. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura. Blood 2002; 99: 3872–3873.
- 48. Patel VL, Mahévas M, Lee SY, et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012; 119: 5989–5995.
- 49. Bussel JB, Lee CS, Seery C, et al. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica 2014; 99: 1264–1271.
- 50. Marangon M, Vianelli N, Palandri F, et al. Rituximab in immune thrombocytopenia: gender, age, and response as predictors of long‐term response. Eur J Haematol 2017; 98: 371–377.
- 51. Mahévas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013; 88: 858–861.
- 52. Zaja F, Vianelli N, Volpetti S, et al. Low‐dose rituximab in adult patients with primary immune thrombocytopenia: low‐dose rituximab in ITP. Eur J Haematol 2010; 85: 329–334.
- 53. Li Y, Shi Y, He Z, et al. The efficacy and safety of low‐dose rituximab in immune thrombocytopenia: a systematic review and meta‐analysis. Platelets 2019; 30: 690–697.
- 54. Hasan A, Michel M, Patel V, et al. Repeated courses of rituximab in chronic ITP: three different regimens. Am J Hematol 2009; 84: 661–665.
- 55. Dysart C, Rozenberg‐Ben‐Dror K, Sales M. Assessing hepatitis B reactivation risk with rituximab and recent intravenous immunoglobulin therapy. Open Forum Infect Dis 2020; 7: ofaa080.
- 56. Nazi I, Kelton JG, Larché M, et al. The effect of rituximab on vaccine responses in patients with immune thrombocytopenia. Blood 2013; 122: 1946–1953.
- 57. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double‐blind, placebo‐controlled trial. Lancet 2009; 373: 641–648.
- 58. Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double‐blind randomised controlled trial. Lancet 2008; 371: 395–403.
- 59. Kuter DJ, Bussel JB, Newland A, et al. Long‐term treatment with romiplostim in patients with chronic immune thrombocytopenia: safety and efficacy. Br J Haematol 2013; 161: 411–423.
- 60. Wong RSM, Saleh MN, Khelif A, et al. Safety and efficacy of long‐term treatment of chronic/persistent ITP with eltrombopag: final results of the EXTEND study. Blood 2017; 130: 2527–2536.
- 61. Ghanima W, Cooper N, Rodeghiero F, et al. Thrombopoietin receptor agonists: ten years later. Haematologica 2019; 104: 1112–1123.
- 62. Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363: 1889–1899.
- 63. Hayes S, Ouellet D, Zhang J, et al. Population PK/PD modeling of eltrombopag in healthy volunteers and patients with immune thrombocytopenic purpura and optimization of response‐guided dosing. J Clin Pharmacol 2011; 51: 1403–1417.
- 64. Quiquandon I, Fenaux P, Caulier MT, et al. Re‐evaluation of the role of azathioprine in the treatment of adult chronic idiopathic thrombocytopenic purpura: a report on 53 cases. Br J Haematol 1990; 74: 223–228.
- 65. Alstead EM, Ritchie JK, Lennard‐Jones JE, et al. Safety of azathioprine in pregnancy in inflammatory bowel disease. Gastroenterology 1990; 99: 443–446.
- 66. Taylor A, Neave L, Solanki S, et al. Mycophenolate mofetil therapy for severe immune thrombocytopenia. Br J Haematol 2015; 171: 625–630.
- 67. Khellaf M, Chabrol A, Mahevas M, et al. Hydroxychloroquine is a good second‐line treatment for adults with immune thrombocytopenia and positive antinuclear antibodies: hydroxychloroquine is a good second‐line treatment for adults. Am J Hematol 2014; 89: 194–198.
- 68. Cuker A, Neunert CE. How I treat refractory immune thrombocytopenia. Blood 2016; 128: 1547–1554.
- 69. Hill QA. How does dapsone work in immune thrombocytopenia? Implications for dosing. Blood 2015; 125: 3666–3668.
- 70. Emilia G, Luppi M, Morselli M, et al. A possible role for low‐dose cyclosporine in refractory immune thrombocytopenic purpura. Haematologica 2008; 93: 1113–1115.
- 71. Mahévas M, Fain O, Ebbo M, et al. The temporary use of thrombopoietin‐receptor agonists may induce a prolonged remission in adult chronic immune thrombocytopenia. Results of a French observational study. Br J Haematol 2014; 165: 865–869.
- 72. Reddy D, Murphy SJ, Kane SV, et al. Relapses of inflammatory bowel disease during pregnancy: in‐hospital management and birth outcomes. Am J Gastroenterol 2008; 103: 1203–1209.
- 73. Khellaf M, Michel M, Quittet P, et al. Romiplostim safety and efficacy for immune thrombocytopenia in clinical practice: 2‐year results of 72 adults in a romiplostim compassionate‐use program. Blood 2011; 118: 4338–4345.
- 74. Nguyen TT, Palmaro A, Montastruc F, et al. Signal for thrombosis with eltrombopag and romiplostim: a disproportionality analysis of spontaneous reports within VigiBase. Drug Saf 2015; 38: 1179–1186.
- 75. Alvarez Blanco JM, Sierra Salazar A, Rangel‐Patiño J, Demichelis R. Search for and extraction of accessory spleen in refractory immune cytopenias. effective or a waste in resources? Blood 2020; 136: 10–11.
- 76. Bradbury CA, Greenwood R, Pell J, et al. A multicentre randomised trial of first line treatment pathways for newly diagnosed immune thrombocytopenia: standard steroid treatment versus combined steroid and mycophenolate — the FLIGHT trial [abstract]. Blood 2020; 136 (Suppl): LBA‐2.
- 77. Estève C, Samson M, Guilhem A, et al. Efficacy and safety of dapsone as second line therapy for adult immune thrombocytopenia: a retrospective study of 42 patients. PLoS One 2017; 12: e0187296.
- 78. Crickx E, Moulis G, Ruivard M, et al. Efficacy and safety of a combination of thrombopoietin receptor agonist with an immunosuppressant therapy for the management of multirefractory adult ITP: results from a retrospective, multicenter, observational study. Blood 2020; 136: 12–13.
- 79. eTG Complete. Primary prophylaxis in immunocompromised adults without HIV infection [website]. Melbourne: Therapeutic Guidelines Limited; 2019. https://www.tg.org.au (viewed Dec 2020).
- 80. Guidry JA, George JN, Vesely SK, et al. Corticosteroid side‐effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83: 175–182.
- 81. Hill QA, Newland AC. Fatigue in immune thrombocytopenia. Br J Haematol 2015; 170: 141–149.
- 82. Neunert C, Noroozi N, Norman G, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost 2015; 13: 457–464.
- 83. Frederiksen H, Maegbaek ML, Nørgaard M. Twenty‐year mortality of adult patients with primary immune thrombocytopenia: a Danish population‐based cohort study. Br J Haematol 2014; 166: 260–267.
- 84. Neunert CE, Buchanan GR, Blanchette V, et al. Relationships among bleeding severity, health‐related quality of life, and platelet count in children with immune thrombocytopenic purpura. Pediatr Blood Cancer 2009; 53: 652–654.
- 85. Choi PY‐I, Roncolato F, Badoux X, et al. A novel triple therapy for ITP using high‐dose dexamethasone, low‐dose rituximab, and cyclosporine (TT4). Blood 2015; 126: 500–503.
- 86. Mayer B, Salama A. Successful treatment of bleeding with tranexamic acid in a series of 12 patients with immune thrombocytopenia. Vox Sanguinis 2017; 112: 767–772.
- 87. Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79: 504–522.
- 88. Spahr JE, Rodgers GM. Treatment of immune‐mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83: 122–125.
- 89. Carr JM, Kruskall MS, Kaye JA, Robinson SH. Efficacy of platelet transfusions in immune thrombocytopenia. Am J Med 1986; 80: 1051–1054.
- 90. Cervantes F, Montserrat E, Rozman C, et al. Low‐dose vincristine in the treatment of corticosteroid‐refractory idiopathic thrombocytopenic purpura (ITP) in non‐splenectomized patients. Postgrad Med J 1980; 56: 711–714.
- 91. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence‐based practice guideline for immune thrombocytopenia. Blood 2011; 117: 4190–4207.
- 92. Harker LA, Finch CA. Thrombokinetics in man. J Clin Invest 1969; 48: 963–974.
- 93. Myers B. Diagnosis and management of maternal thrombocytopenia in pregnancy. Br J Haematol 2012; 158: 3–15.
- 94. Arnold DM, Heddle NM, Cook RJ, et al. Perioperative oral eltrombopag versus intravenous immunoglobulin in patients with immune thrombocytopenia: a non‐inferiority, multicentre, randomised trial. Lancet Haematol 2020; 7: e640–e648.
- 95. Eslick R, Cutts B, Merriman E, et al. HOW Collaborative position paper on the management of thrombocytopenia in pregnancy. Aust N Z J Obstet Gynaecol 2021; 61: 195–204.
- 96. Abrahamson PE, Hall SA, Feudjo‐Tepie M, et al. The incidence of idiopathic thrombocytopenic purpura among adults: a population‐based study and literature review. Eur J Haematol 2009; 83: 83–89.
- 97. Chandan JS, Thomas T, Lee S, et al. The association between idiopathic thrombocytopenic purpura and cardiovascular disease: a retrospective cohort study. J Thromb Haemost 2018; 16: 474–480.
- 98. Fontana V, Jy W, Ahn ER, et al. Increased procoagulant cell‐derived microparticles (C‐MP) in splenectomized patients with ITP. Thromb Res 2008; 122: 599–603.
- 99. Marie I, Maurey G, Hervé F, et al. Intravenous immunoglobulin‐associated arterial and venous thrombosis; report of a series and review of the literature. Br J Dermatol 2006; 155: 714–721.
- 100. Rodeghiero F. ITP and thrombosis: an intriguing association. Blood Adv 2017; 1: 2280.
- 101. Napolitano M, Saccullo G, Marietta M, et al. Platelet cut‐off for anticoagulant therapy in thrombocytopenic patients with blood cancer and venous thromboembolism: an expert consensus. Blood Transfus 2019; 17: 171–180.
- 102. Samuelson Bannow BT, Lee A, Khorana AA, et al. Management of cancer‐associated thrombosis in patients with thrombocytopenia: guidance from the SSC of the ISTH. J Thromb Haemost 2018; 16: 1246–1249.
- 103. Tufano A, Guida A, Di Minno MN, et al. Prevention of venous thromboembolism in medical patients with thrombocytopenia or with platelet dysfunction: a review of the literature. Semin Thromb Hemost 2011; 37: 267–274.
- 104. Newland AC, Sánchez‐González B, Rejtő L, et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am J Hematol 2020; 95: 178–187.
- 105. Bussel J, Arnold DM, Grossbard E, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo‐controlled trials. Am J Hematol 2018; 93: 921–930.
- 106. Kuter DJ, Boccia RV, Lee E‐J, et al. Phase I/II, Open‐label, adaptive study of oral Bruton tyrosine kinase inhibitor PRN1008 in patients with relapsed/refractory primary or secondary immune thrombocytopenia. Blood 2019; 134: 87–87.
- 107. Broome CM, Röth A, Kuter DJ, et al. inhibition of the classical pathway of complement with sutimlimab in chronic immune thrombocytopenic purpura patients without adequate response to two or more prior therapies. Blood 2019; 134: 898–898.
Abstract
Introduction: The absence of high quality evidence for basic clinical dilemmas in immune thrombocytopenic purpura (ITP) underlines the need for contemporary guidelines relevant to the local treatment context. ITP is diagnosed by exclusions, with a hallmark laboratory finding of isolated thrombocytopenia.
Main recommendations: Bleeding, family and medication histories and a review of historical investigations are required to gauge the bleeding risk and possible hereditary syndromes. Beyond the platelet count, the decision to treat is affected by individual bleeding risk, disease stage, side effects of treatment, concomitant medications, and patient preference. Treatment is aimed at achieving a platelet count > 20 × 109/L, and avoidance of severe bleeding. Steroids are the standard first line treatment, with either 6‐week courses of tapering prednisone or repeated courses of high dose dexamethasone providing equivalent efficacy. Intravenous immunoglobulin can be used periprocedurally or as first line therapy in combination with steroids.
Changes in management as a result of this statement: There is no consensus on choice of second line treatments. Options with the most robust evidence include splenectomy, rituximab and thrombopoietin receptor agonists. Other therapies include azathioprine, mycophenolate mofetil, dapsone and vinca alkaloids. Given that up to one‐third of patients achieve a satisfactory haemostatic response, splenectomy should be delayed for at least 12 months if possible. In life‐threatening bleeding, we recommend platelet transfusions to achieve haemostasis, along with intravenous immunoglobulin and high dose steroids.