




New drugs and medical devices offer improvements in health care. Clinical research is undertaken to elucidate such benefits, but also to identify potential harms. Adverse event (AE) and serious AE (SAE) data are crucial information in drug and device development studies (Box 1). Definitions and requirements for safety reporting in Australia are outlined by the international guidelines,1 and the National Health and Medical Research Council (NHMRC) National statement on ethical conduct in human research.2 Further, the NHMRC has provided clarification on how AEs should be reported and who has responsibility for reviewing and acting on them.3 Such guidelines are practical steps to ensure safety for participants in all research involving interventions, including post-marketing surveillance and Phase IV trials (Box 2) of approved medicines, treatments and devices.
However, there is quite marked variation in how, to whom and indeed if such safety reporting is required in investigator-initiated clinical trials. Any such requirement needs to take into account the context of the study, including its phase, the existing health conditions of the participants, and the risks of the proposed intervention over and above standard care. Moreover, the burden and cost of reporting should be proportionate with the public health benefit of this information and be closely linked to a clear need to establish a safety profile, as opposed to simply fulfilling a perceived reporting requirement.
Unfortunately, in Australia there are ever-increasing complexities and burdensome requirements from sponsors and human research ethics committees (HRECs) to report everything without necessarily paying heed to the principles outlined in the guidance documents. The increased burden on investigators and research coordinators is increasing the workload and costs of clinical trials,4 and decreasing opportunities for trial recruitment and participation of centres at the local level. This has led to a decrease in the uptake of research, particularly less well funded investigator-initiated trials.5
We, and others,6,7 believe that individual AE reports to HRECs serve no useful purpose because in most cases the study group identity (drug exposure) is not known, and in any case their value is limited (being a single case report). Further, such reporting is not explicitly mandated by existing guidelines; it is only an option that an HREC may decide to require. In a recent public consultation of safety reporting in Australia, the NHMRC identified several areas for potential reform but did not make specific reference to the issues arising in investigator-initiated studies employing comparisons of approved interventions.8 The purpose of this commentary is to draw attention to these issues and to present some solutions.
Post-marketing surveillance and Phase IV pragmatic drug trials
Although safety reporting is essential for investigational drugs and devices when they are being used for the first time or in a novel indication (Phase I and II, and some Phase III studies), the manner in which they are collected, reported and reviewed should always be proportionate to the size, complexity and risk of the proposed use.9 In situations in which they are being used within their approved indication but may be delivered in the setting of a pragmatic Phase IV drug trial, it can be argued that the threshold for reporting should be commensurate with that expected of their use in routine clinical practice. That is, as all health care professionals are expected to report adverse reactions to medicines through national reporting systems,10-13 we contend that this system should also guide appropriate safety reporting for Phase IV trials.
Known events, as listed in the product information document, need not be reported as AE reporting in post-marketing Phase IV trials. Known AEs should be compiled in an appendix to the study protocol so that investigators and research staff will know of these facts.
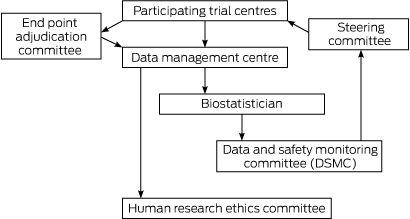
By defining safety endpoints in a study protocol, appropriate data can be collated and reviewed for safety and efficacy by a properly constituted trial data safety and monitoring committee (DSMC). Therefore data review can occur in a meaningful way, by independent experts, with blinded separation of treatment groups enabling useful comparisons for the whole dataset. This then will enable the generation of a concise and relevant safety report for dissemination and review by all participating HRECs. This will relieve overburdened HRECs from having to deal with large numbers of reports which contain minimal information relevant to the true safety of the product under investigation.
Reporting requirements in the United States and the European Union
In Australia, it has been our experience that some HRECs insist on blanket reporting of any event (expected or unexpected, severe or otherwise, treatment-related or otherwise) during a predefined period, using a stringent interpretation of NHMRC guidelines.14 The US Food and Drug Administration (FDA) issued updated guidelines in 2009,15 stating that AEs listed in the investigator’s brochure would, by definition, not be considered unexpected and thus do not require reporting to HRECs. The European Union has adopted similar guidelines.12 That is, an AE should only be reported if the site investigator would be prepared to report the event as part of routine clinical care.
An Australian solution: defining what should be reported
The role of the DSMC is to assess, at predefined intervals, the progress of a clinical trial, safety data and critical efficacy variables, and to recommend to the sponsor whether to continue, modify or terminate a trial.16,17 It is the responsibility of the DSMC to safeguard the individual trial subjects while enhancing the integrity of the trial.18 A DSMC should be made up of individuals who are independent from the trial, but with an understanding of the trial and its components.19 The steering committee of the trial should produce a charter or operating procedures for the DSMC before commencement of the trial,22 and this document should outline the defined role and reporting pathways of the DSMC.
An important role of an HREC is to ensure, with few exceptions, that a clinical trial protocol requires safety (and other) data to be reviewed by a DSMC. In the US, the Office for Human Research Protections and the FDA have agreed that such duplicate reporting (to a DSMC and an institutional review board [IRB; equivalent to an Australian HREC]) is not required,20 and that it has the effect of “inhibiting, rather than enhancing, the ability of IRBs to adequately protect human subjects”.15
The DSMC is in the unique position of being able to review safety information according to the blinded, and sometimes unblinded, treatment arms of the study; that is, it can view the safety data by group and also by site.21 The DSMC can then provide a summary report to the trial steering committee, principal investigators, and in turn the relevant HRECs in a meaningful way (Box 3).
What should be reported?
For Phase IV trials, universal safety endpoints should be routinely collected,16,21 reducing the administrative and AE reporting burden as well as enhance vigilance. The methods used to document these safety endpoints should be described in the protocol, and the data analysed at appropriate time points as determined by the risk profile of the intervention. The International Council for Harmonisation (ICH) guidelines for good clinical practice have identified such universal safety points.12 Universal safety endpoints should include death, readmission or admission to hospital, and return to surgery (if relevant) — these being the major concerns for increased risk to patients, clinicians, HRECs and regulatory bodies. Data collection and AE reporting need to be more meticulous if a study is primarily designed to specifically evaluate safety.
In line with the ICH guidelines12 adopted by the regulatory bodies of the EU, Japan and the US, reportable safety data should be related to drug reactions deemed serious and unexpected. This is consistent with the recommendations of the Council for International Organizations of Medical Sciences,22 which provides essential risk management components. Therefore, reportable events should be defined by:
A single occurrence of a serious, unexpected event that is uncommon and strongly associated with drug exposure (eg, angioedema, agranulocytosis, Stevens-Johnson syndrome).
A single occurrence, or more often a small number of occurrences, of a serious, unexpected event that is not commonly associated with drug exposure, but uncommon in the study population (eg, tendon rupture, progressive multifocal leukoencephalopathy).
AEs should be collected and classified enabling identification of relevant signals in the study, according to treatment group.
An AE that is described or addressed in the investigator’s brochure, protocol, or informed consent documents, but occurs at a specificity or severity that is inconsistent with prior observations (eg, if transaminase elevation is listed in the investigator’s brochure and hepatic necrosis is observed in trial subjects, hepatic necrosis would be considered a reportable AE).15
Conclusions and recommendations
Pragmatic, investigator-initiated Phase IV clinical trials of post-marketed drugs or devices play a critical part in understanding their role in the “real world” of everyday clinical practice.13 In this setting, the workload and costs of systematic, complete reporting of all AEs and SAEs (independent of whether these are treatment-related) is wasteful and mostly unnecessary. There needs to be consistency and relevance applied to SAE reporting in clinical trials.
Definitions of safety events, inclusion of expected trial events for the population under study, interim analyses, DSMC review and a statistical analysis plan should be detailed in the protocol for Phase IV clinical trials.
Defined trial endpoints do not need to be reported as safety events because they are being properly monitored and analysed. Commonly reported SAEs such as events leading to significantly increased hospital length of stay, readmission to hospital or death should be mandated as endpoints in all Phase IV clinical trials.
To reassure HRECs, study participants and the general public, good clinical practice training and certification should be mandated when accepting the role of principal investigator in any clinical trial.
Box 1 – Types of adverse advents
Adverse event (AE): An AE is any untoward medical occurrence in a patient administered a medicinal product which does not necessarily have to have a causal relationship with this treatment. An AE can therefore be any unfavourable and unintended sign (eg, an abnormal laboratory finding), symptom or disease temporally associated with the use of a medicinal product, whether or not considered related to this medicinal product.
Serious adverse event (SAE): An AE is serious and should be reported if it results in death, is life-threatening, requires inpatient hospitalisation or results in prolongation of existing hospitalisation, results in persistent or significant disability/incapacity, is a congenital anomaly/birth defect, or is a medically important event or reaction that may require medical or surgical intervention to prevent one of the other outcomes.
Suspected unexpected serious adverse reactions (SUSARs) and unanticipated serious adverse device effects (USADEs): A serious adverse event with a possible relationship to the study drug, device or procedure should be considered unexpected or unanticipated if the nature, severity or frequency of that event is not consistent with the information on the investigator's brochure, device information or in the current Australian product information.
Box 2 – Types of clinical trials
Phase I study: A new drug or treatment is evaluated in a small number of participants (usually young, healthy males) for the first time to assess safety, dosage range, and identify any adverse effects. The aim is to quantify pharmacokinetic or other performance characteristics, as well as gain an initial safety profile in humans.
Phase II study: The drug or treatment is given to a larger group of participants (often including those with a specific condition) to assess whether it is effective and to further evaluate safety. The main aim is to establish an appropriate dosage schedule to test in later phase studies.
Phase III study: The drug or treatment is given to large groups of participants with a specific condition, most often in the setting of a double-blind randomised trial, to assess its efficacy and adverse effects in comparison to placebo or other established treatments. Results will inform the drug/device product information document. The aim is to use these data to gain marketing approval for use in clinical practice.
Phase IV study: Further studies are carried out after the drug or treatment has been marketed to gather information on the drug or device's effectiveness in broader populations, and any side effects associated with long term use.
Provenance: Not commissioned; externally peer reviewed.
- Sophie Wallace1
- Paul S Myles1,2
- Nikolajs Zeps3
- John R Zalcberg2
- 1 Alfred Hospital, Melbourne, VIC
- 2 Monash University, Melbourne, VIC
- 3 St John of God Perth Northern Hospitals, Perth, WA
Paul Myles is a National Health and Medical Research Council Practitioner Fellow.
No relevant disclosures.
- 1. ICH Expert Working Group. ICH harmonised tripartite guideline. Clinical safety data management: definitions and standards for expedited reporting E2A. Step 4 version dated 27 October 1994. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2A/Step4/E2A_Guideline.pdf (accessed Feb 2016).
- 2. National Health and Medical Research Council, Australian Research Council, Universities Australia. Australian Code for the Responsible Conduct of Research. Canberra: Australian Government, 2007. https://www.nhmrc.gov.au/_files_nhmrc/publications/attachments/r39_australian_code_responsible_conduct_research_150107.pdf (accessed Aug 2015).
- 3. National Health and Medical Research Council. NHMRC Australian Health Ethics Committee (AHEC) position statement. Monitoring and reporting of safety for clinical trials involving therapeutic products. May 2009. Canberra: NHMRC, 2009. http://www.nhmrc.gov.au/_files_nhmrc/file/health_ethics/hrecs/reference/090609_nhmrc_position_statement.pdf (accessed Aug 2015).
- 4. Eapen ZJ, Vavalle JP, Granger CB, et al. Rescuing clinical trials in the United States and beyond: a call for action. Am Heart J 2013; 165: 837-847.
- 5. McNeil JJ, Nelson MR, Tonkin AM. Public funding of large-scale clinical trials in Australia. Med J Aust 2003; 179: 519-520. <MJA full text>
- 6. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluations and Research, Center for Biologics Evaluation and Research. Guidance for industry and investigators: safety reporting requirements for INDs and BA/BE studies. December 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM227351.pdf (accessed Aug 2015).
- 7. Infectious Diseases Society of America. Grinding to a halt: the effects of the increasing regulatory burden on research and quality improvement efforts. Clin Infect Dis 2009; 49: 328-335.
- 8. T Symons Associates. Response to the consultation on updating arrangements for safety monitoring and reporting of clinical trials in Australia. May 2015. https://www.nhmrc.gov.au/_files_nhmrc/file/research/clinical_trials/clinical_trials_consultation_safety_monitoring_report_2015.pdf (accessed Aug 2015).
- 9. European Medicines Agency, Heads of Medicines Agencies. Guideline on good pharmacovigilance practices (GVP). Module VI – Management and reporting of adverse reactions to medicinal products. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/09/WC500172402.pdf (accessed Feb 2016).
- 10. US Food and Drug Administration. FDA Adverse Event Reporting System (FAERS). http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/default.htm (accessed Feb 2016).
- 11. EudraVigilance Mandatory e-reporting essentials. http://eudravigilance.ema.europa.eu/human/index.asp (accessed Aug 2015).
- 12. ICH Expert Working Group. ICH Harmonised tripartite guideline: development safety update report E2F. Step 4 version dated 17 August 2010. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2F/Step4/E2F_Step_4.pdf (accessed Aug 2015).
- 13. World Alliance for Patient Safety. WHO draft guidelines for adverse event reporting and learning systems. World Health Organization, 2005. http://osp.od.nih.gov/sites/default/files/resources/Reporting_Guidelines.pdf (accessed Aug 2015).
- 14. National Health and Medical Research Council. A guide to the development, implementation and evaluation of clinical practice guidelines. Canberra: NHMRC, 1999. https://www.nhmrc.gov.au/guidelines-publications/cp30 (accessed Aug 2015).
- 15. US Food and Drug Administration. Guidance for clinical investigators, sponsors, and IRBs adverse event reporting to IRBs — improving human subject protection. 2009. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126572.pdf (accessed Aug 2015).
- 16. Bruce J, Russell EM, Mollison J, Krukowski ZH. The measurement and monitoring of surgical adverse events. Health Technol Assess 2001; 5: 1-194.
- 17. Chalmers I, Altman DG, McHaffie H, et al. Data sharing among data monitoring committees and responsibilities to patients and science. Trials 2013; 14: 102.
- 18. Fleming TR, Hennekens CH, Pfeffer MA, DeMets DL. Enhancing trial integrity by protecting the independence of data monitoring committees in clinical trials. J Biopharm Stat 2014; 24: 968-975.
- 19. Korn EL, Freidlin B. Inefficacy interim monitoring procedures in randomized clinical trials: the need to report. Am J Bioethics 2011; 11: 2-10.
- 20. Office for Human Research Protections. Guidance on reviewing and reporting unanticipated problems involving risks to subjects or others and adverse events. 2007. http://osp.od.nih.gov/sites/default/files/resources/advevntguid_0.pdf (accessed Aug 2015).
- 21. Pocock SJ, Gersh BJ. Do current clinical trials meet society’s needs?: a critical review of recent evidence. J Am Coll Cardiol 2014; 64: 1615-1628.
- 22. Council for International Organizations of Medical Sciences. Practical approaches to risk minimisation for medicinal products: report of CIOMS Working Group IX. Geneva: CIOMS, 2014.
Summary