




Phaeochromocytomas are rare neoplasms of the chromaffin cells derived from the embryonic neural crest. They are found in fewer than 0.2% of patients with hypertension.1 Most are derived from the adrenal medulla, but the term is also often used for extra-adrenal tumours of the sympathetic nervous system, which may arise in the abdomen, pelvis, thorax, and neck. Both the clinical and biochemical features of phaeo-chromocytomas result largely from the overproduction of catecholamines. Phaeochromocytoma has been called the “10% tumour”, suggesting that 10% are bilateral, 10% malignant, 10% extra-adrenal, and 10% familial. However, in the past 5 years, advances in our understanding of the genetics of this disease and in clinical research have compelled us to re-evaluate this axiom. We review these new insights, with particular attention to familial syndromes, biochemical and radiographic evaluation, and surgical and adjuvant therapy.
It has been known for some time that phaeochromocytomas present as a component of the familial syndromes multiple endocrine neoplasia type 2, von Hippel–Lindau disease, and, rarely, neurofibromatosis type 1. Germline mutations have been identified in these diseases in, respectively, the proto-oncogene RET, and the tumour suppressor genes VHL and NF12 (Box 1).
More recently, germline mutations in the genes encoding succinate dehydrogenase subunit D (SDHD) and subunit B (SDHB) have been described in patients with phaeochromocytomas.3,4 These genes encode subunits of mitochondrial complex II. The mechanism by which the mutations cause tumours remains to be elucidated. However, hypotheses include mitochondrial-mediated failure of apoptosis,3 or even activation of the angiogenic pathway.
Both maternal and paternal inheritance of SDHB mutations resulting in disease phenotype have been shown in familial phaeochromocytomas.6 Recently, a study of 271 predominantly Polish and German patients with non-syndromic (apparently sporadic) phaeochromocytoma found that 66 (24%) were carrying a germline mutation in RET, VHL, SDHD, or SDHB;7 these patients were more likely to present at a young age than those lacking any of these mutations (mean, 25 versus 44 years), and were also more likely to have multifocal disease and extra-adrenal tumours.
Other investigators have reported the prevalence of familial disease to range from 21% to 30%.8,9 Although all these cohort studies may be affected by selection bias because of geographic limitations, taken together they strongly suggest that germline mutations are responsible for phaeochromocytoma in a familial setting more often than previously estimated. Undoubtedly, the discovery of the involvement of SDH genes in phaeochromocytoma is the major factor driving this trend, as the main technical advance in genetic surveys performed since 2001 has been the inclusion of screening for mutations in these genes. Novel loci that may account for additional familial cases of phaeochromocytoma remain to be characterised.10,11
The clinical implication of these findings is that genetic screening should be offered more widely. In fact, it has been recommended that all patients presenting with apparently sporadic phaeochromocytoma should be offered routine analysis for mutations in SDHB, SDHD, VHL, and RET.7 A positive result should prompt biochemical testing and imaging studies (discussed below), as well as the offer of genetic testing and counselling to all first-degree relatives of the patient.
In the past, phaeochromocytoma has been described to present with a characteristic triad of headache, diaphoresis, and palpitations.12 It remains true that the presence of one or more of these paroxysmal symptoms in association with hypertension should alert the physician. However, the era of such classic presentations may be waning, as an increasing proportion of phaeochromocytomas are detected at an early stage, before clinical signs and symptoms become apparent. This may be attributable to more widespread application of genetic testing as more patients present with subclinical disease (eg, asymptomatic catecholamine excess) in the setting of a strong family history and with abnormalities on abdominal imaging studies.
Clinically inapparent adrenal masses, or “incidentalomas”, are common in the general population, found in 2.1% of autopsies and 1%–4% of abdominal imaging studies.13 Fewer than 10% of such lesions are subsequently determined to be phaeochromocytomas. Conversely, in a recent study of 39 consecutive patients undergoing laparoscopic adrenalectomy for phaeochromocytoma at a single centre, 17 patients (44%) had initially presented with adrenal incidentalomas.14 A US National Institutes of Health expert panel in 2003 recommended biochemical evaluation of all patients with incidentally discovered adrenal masses, and surgical removal of lesions proven to hypersecrete catecholamines.13
Measurements of urinary catecholamines and their metabolites, metanephrines, over 24 hours have been the mainstay of biochemical diagnosis of phaeochromocytoma for many years. These tests are widely available and have been confirmed to be 99% specific when appropriate cut-off values are used (roughly twice the upper limit of the reference range).15 When measurements of urinary catecholamines and their metabolites are considered as a grouped test (positive when levels of any single compound are elevated), an acceptable sensitivity of 88% is achieved.
However, a large multicentre cohort study in 2002 found that measurement of plasma levels of free metanephrines by high-pressure liquid chromatography detected phaeochromocytomas with a sensitivity of 99%.16 The advantage of assaying metanephrines is that they are produced continuously, resulting in steady-state levels. In contrast, catecholamines are released episodically. The high sensitivity of this test has been confirmed by two other studies, and there is growing consensus that a negative result on this single test is sufficient to exclude phaeochromocytoma.17
Difficulty arises when plasma levels of free metanephrines are raised, as the specificity is only 85%. Given the low prevalence of phaeochromocytoma, it is estimated that 30 people have false-positive results for every one patient with phaeochromocytoma identified.15 A number of drugs and medical conditions (most notably congestive cardiac failure, major depression and panic disorder) are known to confound measurement of both plasma and urinary catecholamines and their metabolites.18 In a recent study, tricyclic antidepressants and phenoxybenzamine accounted for 45% of false-positive elevations of plasma nor-epinephrine and normetanephrine levels.19 This report also demonstrated that clonidine suppression testing (measurement of plasma free normetanephrine level after oral administration of 0.3 mg clonidine) may help clarify equivocal test results. Another major drawback of plasma free metanephrine testing is its limited availability in comparison with urine testing.
Other tests, such as catecholamine stimulation testing with glucagon, histamine, tyramine or naloxone, are largely obsolete.
In general, imaging studies are justified only after the diagnosis of phaeochromocytoma has been established biochemically. Currently available imaging methods for localisation of phaeochromocytoma can be grouped into anatomic modalities (computed tomography [CT] and magnetic resonance imaging [MRI]) and functional imaging modalities (scintigraphy). CT and MRI (Box 2A) have a sensitivity of 90%–95%, with MRI being superior for detecting extra-adrenal tumours. However, both offer poor specificity (as low as 50% in some reports).20
The most commonly used functional study for detecting phaeochromocytoma is scintigraphy with 131I-labelled metaiodobenzylguanidine (131I-MIBG), which has specificity of over 95% but sensitivity of only 77%–90%.21 Scintigraphy using MIBG labelled with 123I instead of 131I is reported to yield superior images, but the short half-life of this isotope (13.2 hours) limits its use to metropolitan centres with a nearby cyclotron (Box 2B).
Since 1990, five novel functional imaging techniques have been developed and compared with established methods. These comprise somatostatin receptor scintigraphy with 111In-pentetreotide, and positron emission tomography (PET) using 18F-fluorodeoxyglucose, 11C-hydroxyephedrine, 18F-dihydroxyphenylalanine, and most recently 6-18F-fluorodopamine.21 Although none of these techniques is in clinical use to date, PET with 6-18F-fluorodopamine was found to have 100% sensitivity in the detection of metastatic phaeochromocytoma in a recent study of 16 patients.22 Seven of these patients had negative 131I-MIBG scans, and the technique detected substantially more metastatic foci than 131I-MIBG scanning.
Surgical excision remains the cornerstone of treatment for phaeochromocytoma, achieving cure in more than 90% of cases. An audit of surgery for phaeochromocytoma at a single centre over 23 years found that medical and surgical advances have greatly reduced adverse perioperative events.9 The overall mortality rate was 2.4%. Non-fatal complications occurred in 38 of 165 operations (involving 147 patients), most notably splenic injuries requiring splenectomy in 13 patients. The authors attributed this primarily to the surgical technique used (open bilateral exploration), as only three splenic complications occurred during the last 10 years of the study, when focused exploration was generally used. Complications were most strongly associated with repeat surgery, although high preoperative systolic blood pressure and high urinary metanephrine secretion were also independent risk factors.
Preoperative patient preparation with α-adrenergic blockade (commonly achieved with prazosin or phenoxybenzamine) and volume expansion have accounted for the most significant reductions in perioperative mortality.23 Improved localising studies have enabled focused adrenalectomy. Laparoscopic adrenalectomy has become the procedure of choice for most patients (Box 3), with open adrenalectomy generally reserved for large tumours or those known preoperatively to be malignant. Although large tumours are more likely to be malignant, the upper limit of size appropriate for laparoscopic resection remains at the discretion of the individual surgeon.24
It is generally accepted that about 10% of phaeochromocytomas are malignant. However, we now know that certain subgroups of patients are at particularly high risk. A recent comparison of SDHB and SDHD mutation carriers showed that SDHB carriers were significantly more likely to have malignant tumours, as defined by the identification of tumours in the lungs or bones.5
Despite attempts to establish histopathological criteria to distinguish benign from malignant phaeochromocytomas,25 malignancy remains best defined by the presence of metastases. Metastatic implants are detected by imaging studies demonstrating neoplasia at sites distant from the primary tumour, where neuroectodermal tissues are not normally found.
Gene expression analysis has recently raised the possibility that molecular markers might predict clinical tumour behaviour.
Human telomerase reverse transcriptase is more commonly expressed in malignant phaeochromocytomas compared with benign phaeochromocytomas.26 A number of other molecular markers, including proangiogenic factors, have been examined in preclinical studies in the past 5 years.10 Expression analysis using a matrix of genes known to promote tumour progression may eventually allow for accurate determination of prognosis.
Phaeochromocytomas are generally radioresistant, and chemotherapy most often produces only temporary improvement. Thus, the management of malignant phaeochromocytoma has centred on palliation: medical control of hypertension, surgical debulking, and combination chemotherapy.27 Since the 1980s, some specialised centres have treated patients with high-dose 131I-MIBG radionuclide therapy,28 with overall initial improvement in symptoms (75%), hormonal response (45%) and tumour size (30%). However, long-term benefit is rare, and tumour recrudescence is usual. In 1999, a study in six patients showed that bimodal therapy with 131I-MIBG and combination cytotoxic chemotherapy produced additive effects in reducing tumour volume and function.29
Phaeochromocytoma remains a challenging entity to diagnose and to treat. In the future, genetic screening of at-risk individuals should identify an increasing number of patients with early stage disease. Biochemical testing of plasma or urinary analytes should be used for initial screening of patients with hypertension and suggestive features. After appropriate tumour localisation, most patients with phaeochromocytoma can be treated by minimally invasive techniques. We await further advances from preclinical studies of the diagnosis and treatment of malignant phaeochromocytoma.
1 Gene mutations associated with familial phaeochromocytoma3-5
Gene |
Syndrome |
Other manifestations |
Lifetime risk of tumour (%) |
||||||||||||
RET |
Multiple endocrine neoplasia type 2A |
Medullary thyroid carcinoma, hyperparathyroidism |
40% |
||||||||||||
RET |
Multiple endocrine neoplasia type 2B |
Medullary thyroid carcinoma, mucosal (oral) neuromas, intestinal ganglioneuromas, marfanoid habitus |
40% |
||||||||||||
VHL |
Von Hippel–Lindau disease |
Multiple infratentorial haemangioblastomas and retinal angiomas, renal cell (clear cell) carcinoma, renal and pancreatic cysts, papillary cystadenomas of the reproductive tract and ear |
10%–20% |
||||||||||||
NF1 |
Neurofibromatosis type 1 |
Peripheral neurofibromas, café-au-lait spots, intertriginous freckling, Lisch nodules, optic gliomas, bony abnormalities, other CNS tumours |
< 5% |
||||||||||||
SDHD |
Familial phaeochromocytoma/ paraganglioma |
None identified |
Unknown |
||||||||||||
SDHB |
Familial phaeochromocytoma/ paraganglioma |
Possibly renal cell carcinoma |
Unknown |
||||||||||||
SDHD = succinate dehydrogenase subunit D. SDHB = succinate dehydrogenase subunit B. CNS = central nervous system. |
|||||||||||||||
2 Radiographic findings in phaeochromocytoma
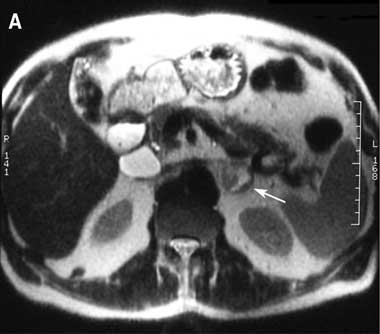

A Magnetic resonance imaging (T2-weighted) showing left adrenal mass with high signal intensity (arrow).
B Whole-body scintigraphy with 123l-metaiodobenzylguanidine, showing focal radiotracer uptake indicative of a left extra-adrenal phaeochromocytoma (side of uptake is inferior to the usual position of the adrenal gland).


- 1. Pacak K, Linehan WM, Eisenhofer G, et al. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 2001; 134: 315-329.
- 2. Dluhy RG. Pheochromocytoma — death of an axiom. N Engl J Med 2002; 346: 1486-1488.
- 3. Astuti D, Douglas F, Lennard TW, et al. Germline SDHD mutation in familial phaeochromocytoma. Lancet 2001; 357: 1181-1182.
- 4. Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 2001; 69: 49-54.
- 5. Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004; 292: 943-951.
- 6. Benn DE, Croxson MS, Tucker K, et al. Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas. Oncogene 2003; 22: 1358-1364.
- 7. Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346: 1459-1466.
- 8. Opocher G, Schiavi F, Conton P, et al. Clinical and genetic aspects of phaeochromocytoma. Horm Res 2003; 59 Suppl 1: 56-61.
- 9. Plouin PF, Duclos JM, Soppelsa F, et al. Factors associated with peri-operative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab 2001; 86: 1480-1486.
- 10. Eisenhofer G, Bornstein SR, Brouwers FM, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer 2004; 11: 423-436.
- 11. Opocher G, Schiavi F, Vettori A, et al. Fine analysis of the short arm of chromosome 1 in sporadic and familial pheochromocytoma. Clin Endocrinol (Oxf) 2003; 59: 707-715.
- 12. Pommier RF, Brennan MF. Management of adrenal neoplasms. Curr Probl Surg 1991; 28: 657-739.
- 13. Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann Intern Med 2003; 138: 424-429.
- 14. Cheah WK, Clark OH, Horn JK, et al. Laparoscopic adrenalectomy for pheochromocytoma. World J Surg 2002; 26: 1048-1051.
- 15. Kudva YC, Sawka AM, Young WF Jr. Clinical review 164: the laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic experience. J Clin Endocrinol Metab 2003; 88: 4533-4539.
- 16. Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002; 287: 1427-1434.
- 17. Sawka AM, Prebtani AP, Thabane L, et al. A systematic review of the literature examining the diagnostic efficacy of measurement of fractionated plasma free metanephrines in the biochemical diagnosis of pheochromocytoma. BMC Endocr Disord 2004; 4: 2.
- 18. Harding JL, Yeh MW, Coleman MJ, et al. Potential pitfalls in the diagnosis of phaeochromocytoma. Med J Aust 2005; 182: 637-639. <MJA full text>
- 19. Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab 2003; 88: 2656-2666.
- 20. Jalil ND, Pattou FN, Combemale F, et al. Effectiveness and limits of preoperative imaging studies for the localisation of pheochromocytomas and paragangliomas: a review of 282 cases. French Association of Surgery (AFC), and The French Association of Endocrine Surgeons (AFCE). Eur J Surg 1998; 164: 23-28.
- 21. Ilias I, Pacak K. Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab 2004; 89: 479-491.
- 22. Ilias I, Yu J, Carrasquillo JA, et al. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab 2003; 88: 4083-4087.
- 23. Duh QY. Evolving surgical management for patients with pheochromocytoma. J Clin Endocrinol Metab 2001; 86: 1477-1479.
- 24. Shen WT, Sturgeon C, Clark OH, et al. Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas. Surgery 2004; 136: 1129-1137.
- 25. Thompson LD. Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002; 26: 551-566.
- 26. Elder EE, Xu D, Hoog A, et al. KI-67 AND hTERT expression can aid in the distinction between malignant and benign pheochromocytoma and paraganglioma. Mod Pathol 2003; 16: 246-255.
- 27. Edstrom Elder E, Hjelm Skog AL, Hoog A, Hamberger B. The management of benign and malignant pheochromocytoma and abdominal paraganglioma. Eur J Surg Oncol 2003; 29: 278-283.
- 28. Loh KC, Fitzgerald PA, Matthay KK, et al. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest 1997; 20: 648-658.
- 29. Sisson JC, Shapiro B, Shulkin BL, et al. Treatment of malignant pheochromocytomas with 131-I metaiodobenzylguanidine and chemotherapy. Am J Clin Oncol 1999; 22: 364-370.
Abstract
The discovery of novel mutations in genes encoding succinate dehydrogenase subunits has revealed that familial phaeochromocytomas are much more common than previously thought.
Genetic screening should be offered to patients with apparently sporadic phaeochromocytomas and their first-degree relatives.
An increasing proportion of phaeochromocytomas present preclinically on genetic testing or as “incidentalomas” on abdominal imaging, rather than with classic symptoms and signs.
Clinical suspicion should prompt measurement of plasma levels of free metanephrine or 24-hour urinary catecholamine and metanephrine levels, followed, if positive, by tumour localisation studies.
With appropriate perioperative care, surgical management of phaeochromocytomas is safe and effective. Most tumours can be removed laparoscopically.