




Leukaemia accounts for about a third of all childhood malignancies. About 80% of children with leukaemia have acute lymphoblastic leukaemia (ALL), 17% have acute myeloid leukaemia (AML), and the remainder have rare forms of chronic leukaemia.1 Collaborative clinical trials research has brought about dramatic improvements in cure rates for children with acute leukaemia, but about 20% of children still relapse. Most of those who relapse will die of the disease. Moreover, some survivors face an uncertain future of health problems because of treatment side effects. Here we review selected recent developments in childhood leukaemia.
When confronted with the diagnosis of childhood leukaemia, one of the first questions asked by parents is “Why did this happen to our child?”. Unfortunately, the answer to this question largely remains unknown. Although a small proportion of children with leukaemia have an underlying predisposition, such as Down syndrome or Fanconi anaemia (a congenital form of aplastic anaemia), most have no known cause. However, in recent years, a molecular basis of childhood leukaemia has become clearer.
Evidence now supports the concept that, for a substantial number of patients, childhood leukaemia begins as an in-utero event. Much of this evidence arises from studies of monozygotic twins who concordantly develop leukaemia,2-4 and from recent polymerase chain reaction studies of neonatal Guthrie card blood spots. The latter studies have shown that 70% of children presenting with ALL up to 13 years of age had molecular evidence of their disease at birth: preleukaemic cells that were present at birth bore the same molecular markers as their leukaemic cells.5,6
This evidence has led to general consensus that most cases of childhood leukaemia begin in utero, but major questions remain as to what leads to these initial genetic abnormalities. Although no specific causes have been identified which result in these in-utero molecular changes, several antenatal factors are important in increasing the risk of leukaemogenesis. These include maternal and paternal marijuana use both before and during pregnancy, which significantly increases the risk of both AML and ALL in offspring.7,8 It has also been suggested that medications used in pregnancy, such as antihistamines and amphetamines, may increase the risk of ALL.8 A retrospective Western Australian study found that, in children whose mothers took folate during pregnancy, the risk of ALL was reduced by 60%.9 These observations have prompted a prospective Australian, multicentre, case–control study aimed at elucidating the role of folate in pregnancy and other possible environmental and genetic risk factors.
Whatever events initiate the development of preleukaemic cells in the fetus, other genetic changes are needed for the development of overt leukaemia — consistent with the “multi-hit” model of leukaemogenesis.10 One study has shown that 1% of cord blood samples from healthy babies contained molecular evidence of the commonest leukaemia-specific chromosomal translocation seen in ALL: t(12; 21), or the TEL-AML1 fusion gene.11 This incidence of a molecular abnormality is almost 100 times greater than the incidence of ALL, suggesting that a second event must occur in 1% of those children with preleukaemic cells to allow progression to overt leukaemia. The nature of this “second hit” is unknown. Exposure to electromagnetic fields has been ruled out as playing any significant role.12 Other possibilities still being investigated include exposure to chemicals, such as pesticides, and childhood infections.13,14 The hypothesis that more than one hit is required for leukaemogenesis has been supported by recent studies into children with Down syndrome who develop acute megakaryoblastic leukaemia. About 10% of all Down syndrome patients develop a transient myeloproliferative disorder (TMD) in the neonatal period. This is indistinguishable from acute leukaemia, and undergoes spontaneous remission. However, a fifth of patients with TMD subsequently develop acute megakaryoblastic leukaemia.15 Recently, mutations in a gene called GATA1, necessary for normal development of erythroid and megakaryocytic cells, have been found in almost all Down syndrome patients with TMD or acute megakaryoblastic leukaemia.16 This suggests that constitutional trisomy 21, together with the GATA1 mutation, causes the excessive megakaryocyte proliferation seen in TMD, and that another hit is necessary to cause overt leukaemia in a smaller subset of patients.17
One of the great successes of paediatric oncology has been the dramatic improvement in cure rate for ALL in recent decades (Box 1).18 Expected current cure rates approach 80% for all children diagnosed with ALL, of all lineages.1 However, relapse remains a problem for the remaining 20% of patients. Although some children have known indicators of poor prognosis, such as a high initial white cell count or cytogenetic abnormalities, most who relapse do not have any clinical markers to indicate increased risk for treatment failure.
The effectiveness of PCR testing for minimal residual disease (MRD) has been investigated as a predictor for relapse. MRD testing is an extremely sensitive method of detecting leukaemic cells not found with standard techniques in patients who are in remission. Several studies have confirmed that MRD levels are a sensitive prognostic indicator (Box 2).19-21 The Australian and New Zealand Children’s Haematology and Oncology Group is currently enrolling new ALL patients in a pilot treatment protocol, in which patients with high MRD levels will be treated with more intensive chemotherapy and, if there is a suitable donor, with stem cell transplantation. The trial addresses the hypothesis that early, very intensive treatment during first remission will prevent relapse in these high-risk patients.
Most children with ALL will be cured with chemotherapy alone. Although there is no established role for autologous stem cell transplantation in ALL, some patients can be identified who are clear candidates for allogeneic bone marrow transplantation. The 1%–2% of patients with Philadelphia chromosome-positive ALL are, for example, a universally accepted subgroup for whom transplantation in first remission is indicated. A recent international review concluded that transplantation with stem cells from a matched related donor in these patients is clearly superior to treatment with chemotherapy alone.22 Some groups also use stem cell transplantation for other groups of very high risk patients, based on clinical criteria such as older age, initial high white cell count and male sex. The marked increase in the size of marrow donor registries and cord blood banks now means that 70%–80% of children needing a bone marrow transplant in Australia will have a suitable allogeneic stem cell unit available. However, acute transplant-related mortality rates remain high, and militate against the widespread use of transplant in childhood ALL, except in children at high risk of relapse.
With the dramatic improvements in survival of children with ALL over the past few decades, attention has increasingly turned to reducing the late effects of treatment, such as the high incidence of impaired neurocognitive outcomes in children receiving prophylactic cranial irradiation.23 As a consequence of randomised trials comparing intrathecal chemotherapy with radiation for central nervous system prophylaxis, irradiation is now omitted in almost 80% of patients. Intrathecal chemotherapy combined with high dose methotrexate has been shown to be equally effective at preventing meningeal relapse in most patients.24 A large extended follow-up study of survivors of childhood ALL showed that non-irradiated patients had similar employment and marital prospects to the rest of the population.25 That study also found that non-irradiated patients who had survived at least 10 years did not have an increased risk of second malignancy. An earlier, very large cohort study of 8831 patients confirmed that with current treatment protocols the risk of second malignancy in the first 10 years after treatment is now as low as 1.18%.26 This low rate is, in part, because of the avoidance of epipodophyllotoxins such as etoposide, and because of the reduction in radiation dosage.
Cranial irradiation has also been implicated as a cause of growth impairment by causing growth hormone deficiency in children treated for ALL. However, a recent study found that children treated for ALL had some impairment of growth irrespective of treatment modality, with the worst outcomes in younger patients.27 Children with poor growth following treatment for ALL need careful follow-up and endocrinological review.
Anthracyclines, which can be responsible for clinical cardiac toxicity, are used in most treatment protocols for ALL. Although the doses used are low enough to avoid causing acute cardiomyopathy during treatment, it is now apparent that children may be at a greater risk of late-onset cardiac failure than previously suspected.28,29 Attempts to reduce the cardiac toxicity of anthracyclines have, to date, been disappointing. The administration of doxorubicin by continuous infusion has not been found to offer significant cardioprotection.30 The iron chelator dexrazoxane has been shown to have some cardio-protectant effect in anthracycline-based therapies of selected adult malignancies.31 A recent study has also suggested that it is protective in children with ALL, but this needs confirmation with long-term assessment.32 Meanwhile, careful monitoring and follow-up of children at risk of cardiac disease is essential.
Pharmacogenetics is a new field of research in ALL. It examines how genetic differences between individuals affect their responses to drug therapy. Because of the narrow therapeutic range of most drugs used in treating childhood ALL, genetic variations in drug metabolism may have an important effect on treatment outcome.33 6-Mercaptopurine, a key medication in ALL treatment, is inactivated by thiopurine methyltransferase in the liver. Mutations have been identified which result in reduced enzyme activity. ALL patients who are homozygous for the deficiency are at risk of life-threatening toxicity from their inability to metabolise normal doses of 6-mercaptopurine. Such patients require substantial reduction in doses of mercaptopurine. However, heterozygotes have improved overall survival, because they have higher levels of systemic 6-mercaptopurine, which are both tolerable and more effective against leukaemic cells.34 Deficient thiopurine methyltransferase activity has also been associated with significantly greater risk of brain tumours in patients receiving both mercaptopurine and cranial irradiation.33 Similarly, patients with polymorphisms in the gene coding for methylenetetrahydrofolate reductase, an important enzyme in folate metabolism, have an increased risk of toxicity during methotrexate treatment.35
Although it is not yet standard practice to assess individual patients for genetic polymorphisms, our knowledge of interpatient variability in drug metabolism is already affecting clinical practice. For example, it has been demonstrated that individualising dosages of mercaptopurine and methotrexate during maintenance therapy, by increasing them to the maximal tolerated level, can improve survival provided that prolonged neutropenia is avoided.36
There is now substantial evidence that adolescents treated with paediatric protocols for ALL have better outcomes than those treated with adult protocols. In one study, the 6-year event-free survival (EFS) rate was 64% for the paediatric protocol compared with 38% for the adult protocol, and, in another, the 5-year EFS rate was 67%, compared with 41%.37,38 Additionally, adolescents and young adults with ALL treated in adult centres are much less likely to be enrolled in clinical trials than children.39 Adolescents’ needs also differ from those of both children and adults. There are special issues relating to self-image, independence, sexuality, education, career development and compliance with treatment.39 There is now progress in cancer centres in Europe and North America towards establishing specialist “teenage cancer units”, which are able to provide specialised medical, nursing and psychosocial treatments. Such units have yet to be established in Australia. We believe that this needs to be redressed.
The outlook for childhood AML remains less encouraging, with cure rates between 35% and 50%.1 Children treated with Australasian protocols have demonstrated comparatively high overall survival rates of 56%, with disease-free survival of 50%.40 Initial chemotherapy will achieve remission in more than 90% of patients, but is extremely myelosuppressive and requires high quality supportive care. Interestingly, children with Down syndrome and AML have a prognosis for cure of 80%–90% when treated with chemotherapy alone.41
Bone marrow transplantation from a matched sibling donor for patients in first complete remission is now established as the treatment of choice for high-risk AML.42 The cytogenetic profile of the leukaemic cells at diagnosis is important in defining high- and low-risk subgroups. For most children with standard-risk AML in first remission without a fully matched sibling donor, either autologous transplantation or further intensive post-remission chemotherapy is used. Some studies have shown a survival advantage for patients undergoing autologous transplantation. However, several recent studies have shown that intensive chemotherapy is just as effective, with the issue remaining unresolved.43 Unrelated allogeneic transplants result in lower relapse rates but higher treatment-related mortality, making this option suitable for high-risk patients only.
The focus of research in AML has shifted to the development of targeted therapy. The antigen CD33 has been the focus of intensive investigation. This has been an attractive target because it is expressed on most myeloid blasts but is absent from normal hematopoietic stem cells and other tissues. The drug gemtuzumab ozogamicin (Mylotarg, Wyeth) has been developed as an immunoconjugate of an anti-CD33 antibody and calicheamicin — an extremely potent cytotoxic agent that cleaves double-stranded DNA.44 Phase 1 and 2 trials in adults and children with relapsed AML have shown responses in 30% of patients.45 A major international paediatric trial to further test the efficacy of gemtuzumab ozogamicin is now open to Australian patients.
1 Progressive increase in long-term survival for children with acute lymphoblastic leukaemia*
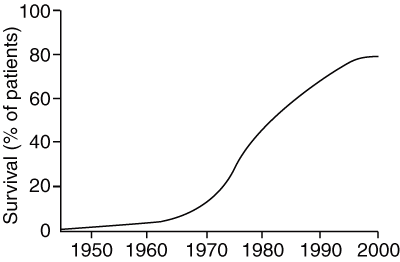
* From Simone JV. Childhood leukemia — successes and challenges for survivors. N Engl J Med 2003; 349: 627-628.18 © 2003 Massachusetts Medical Society. All rights reserved. Reprinted with permission.
2 Minimum residual disease levels after 1 month of treatment correlate strongly with survival in children with acute lymphocytic leukaemia*
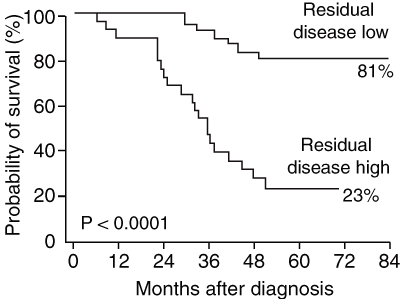
* From Marshall GM, Haber M, Kwan E, et al. Importance of minimal residual disease testing during the second year of therapy for children with acute lymphoblastic leukemia. J Clin Oncol 2003; 21: 704-709.20 Reprinted with permission from the American Society of Clinical Oncology.
- David S Ziegler1
- Glenn M Marshall2
- Luciano Dalla Pozza3
- Keith D Waters4
- 1 Centre for Children’s Cancer and Blood Disorders, Sydney Children’s Hospital, Randwick, NSW.
- 2 Oncology Unit, Children’s Hospital at Westmead, Westmead, NSW.
- 3 Department of Haematology and Oncology, Royal Children’s Hospital, Melbourne, VIC.
None identified.
- 1. Ravindranath Y. Recent advances in pediatric acute lymphoblastic and myeloid leukemia. Curr Opin Oncol 2003; 15: 23-35.
- 2. Wolman IJ. Parallel responses to chemotherapy in identical twins with concordant leukaemia in monozygotic twins. J Pediatr 1962; 60: 91-96.
- 3. Ford AM, Ridge SA, Cabrera ME, et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature 1993; 363: 358-360.
- 4. Wiemels JL, Ford AM, Van Wering ER, et al. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood 1999; 64: 1057-1062.
- 5. Gale KB, Ford AM, Repp R, et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci U S A 1997; 94: 13950-13954.
- 6. Taub JW, Konrad YG, Naber JM, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic lymphoma. Blood 2002; 99: 2992-2996.
- 7. Robison LL, Buckley JD, Daigle AE, et al. Maternal drug use and risk of childhood nonlymphoblastic leukemia among offspring — an epidemiologic investigation implicating marijuana (a report form the Children’s Cancer Study Group). Cancer 1989; 63: 1904-1911.
- 8. Wen W, Shu XO, Potter JD, et al. Parental medication use and risk of childhood acute lymphoblastic leukemia. Cancer 2002; 95: 1786-1794.
- 9. Thompson JR, Gerald PF, Willoughby ML, Armstrong BK. Maternal folate supplementation in pregnancy and protection against acute lymphoblastic leukaemia in childhood: a case–control study. Lancet 2001; 358: 1935-1940.
- 10. Greaves M. Childhood leukaemia. BMJ 2002; 324: 283-287.
- 11. Mori H, Colman SM, Xiao Z, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 2002; 99: 8242-8247.
- 12. UK Childhood Cancer Study Investigators. Childhood cancer and residential proximity to power lines. Br J Cancer 2000; 83: 1573-1580.
- 13. Davies SM, Robison LL, Buckley JD, et al. Glutathione S-transferase polymorphisms in children with myeloid leukemia: a Children’s Cancer Group study. Cancer Epidemiol Biomarkers Prev 2000; 9: 563-566.
- 14. Zahm SH, Ward MH. Pesticides and susceptibility to cancer. Environ Health Perspect 1998; 106(Suppl): 893-908.
- 15. Zipursky A. Transient leukemia — a benign form of leukemia in newborn infants with trisomy 21. Br J Haematol 2003; 120: 930-938.
- 16. Hitzler JK, Cheung J, Li Y, et al. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 2003; 101: 4301-4304.
- 17. Gurbuxani S, Vyas P, Crispino JD. Recent insights into the mechanisms of myeloid leukemogenesis in Down syndrome. Blood 2004; 103: 399-406.
- 18. Simone JV. Childhood leukemia — successes and challenges for survivors. N Engl J Med 2003; 349: 627-628.
- 19. Brisco MJ, Condon J, Hughes E, et al. Outcome prediction in childhood acute lymphoblastic leukaemia by molecular quantification of residual disease at the end of induction. Lancet 1994; 343: 196-200.
- 20. Marshall GM, Haber M, Kwan E, et al. Importance of minimal residual disease testing during the second year of therapy for children with acute lymphoblastic leukemia. J Clin Oncol 2003; 21: 704-709.
- 21. Van Dongen JJM, Seriu T, Panzergrumayer ER, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998; 352: 1731-1738.
- 22. Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med 2000; 342: 998-1006.
- 23. Smibert E, Anderson V, Godber T, Eckert H. Risk factors for intellectual and educational sequelae of cranial irradiation in childhood acute lymphoblastic leukaemia. Br J Cancer 1996; 73: 825-830.
- 24. Hill FG, Richards S, Gibson B, et al. Successful treatment without radiotherapy of children receiving intensified chemotherapy for acute lymphoblastic leukaemia: results of the risk-stratified randomized central nervous system treatment trial MRC UKALL XI. Br J Haematol 2004; 124: 33-46.
- 25. Pui CH, Cheng C, Leung W, et al. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. N Engl J Med 2003; 349: 640-649.
- 26. Bhatia S, Sather HN, Pabustan OB, et al. Low incidence of second neoplasms among children diagnosed with acute lymphoblastic leukemia after 1983. Blood 2002; 99: 4257-4264.
- 27. Dalton VK, Montse R, Silverman LB, et al. Height and weight in children treated for acute lymphoblastic leukemia: relationship to CNS treatment. J Clin Oncol 2003; 21: 2953-2960.
- 28. Sorensen K, Levitt G, Bull C, et al. Anthracycline dose in childhood acute lymphoblastic leukemia: issues of early survival versus late cardiotoxicity. J Clin Oncol 1997; 15: 61-68.
- 29. Silber JH, Cnaan A, Clark BJ, et al. Enalapril to prevent cardiac function decline in long-term survivors of pediatric cancer exposed to anthracyclines. J Clin Oncol 2004; 22: 820-828.
- 30. Lipshultz SE, Giantris AL, Lipsitz SR, et al. Doxorubicin by continuous infusion is not cardioprotective: the Dana-Farber 91-01 acute lymphoblastic leukemia protocol. J Clin Oncol 2002; 20: 1677-1682.
- 31. Schuchter LM, Hensley ML, Meropol NJ, Winer EP. 2002 update of recommendations for the use of chemotherapy and radiotherapy protectants: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol 2002; 20: 2895-2903.
- 32. Lipshutlz SE, Rifai N, Dalton VM, et al. The effect of Dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med 2004; 351: 145-153.
- 33. Carroll WL, Bhojwani D, Min DJ, et al. Pediatric acute lymphoblastic leukemia. Hematology (Am Soc Hematol Educ Program) 2003: 102-131.
- 34. Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91: 2001-2008.
- 35. Chiusolo P, Reddiconto G, Casorelli I, et al. Preponderance of methylenetetrahydrofolate reductase C677T homozygosity among leukemia patients intolerant to methotrexate. Ann Oncol 2002; 13: 1915-1918.
- 36. Relling MV, Hancock ML, Boyett JM, et al. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93: 2817-2823.
- 37. Stock W, Sather H, Dodge RK, et al. Outcome of adolescents and young adults with ALL: A comparison of Children’s Cancer Group (CCG) and Cancer and Leukemia Group B (CALGB) regimens. Blood 2000; 96: 467a.
- 38. Boisel N, Auclerc M-F, Lheritier V, et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 2003; 23: 774-780.
- 39. Albritton K, Bleyer WA. The management of cancer in the older adolescent. Eur J Cancer 2003; 39: 2584-2599.
- 40. O’Brien TA, Russell SJ, Vowels MR, et al. Results of consecutive trials for children newly diagnosed with acute myeloid leukemia from the Australian and New Zealand Children’s Cancer Study Group. Blood 2002; 100: 2708-2716.
- 41. Ravindranath Y. Down syndrome and acute myeloid leukemia: the paradox of increased risk for leukemia and heightened sensitivity to chemotherapy. J Clin Oncol 2003; 21: 3385-3387.
- 42. Woods WG, Neudorf S, Gold S, et al. A comparison of allogeneic bone marrow transplantation, autologous transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood 2001; 97: 56-62.
- 43. Ravindrath Y, Yeager AM, Chang MN, et al. Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. N Engl J Med 1996; 334: 1428-1434.
- 44. Clark JJ, Smith FO, Arceci RJ, et al. Update in childhood acute myeloid leukemia: recent developments in the molecular basis of disease and novel therapies. Curr Opin Hematol 2003; 10: 31-39.
- 45. Zwaan CM, Reinhardt D, Corbacioglu S, et al. Gemtuzumab ozogamicin: first clinical experiences in children with relapsed/refractory acute myeloid leukemia treated on compassionate-use basis. Blood 2003; 101: 3868-2871.
Abstract
In most cases, childhood leukaemia has a fetal origin, but multiple molecular events are required after birth for pre-leukaemic cells to progress to leukaemia.
Cure rates for acute lymphoblastic leukaemia (ALL) now approach 80%.
A high level of minimal residual disease detected by polymerase chain reaction in patients with ALL in remission has profound prognostic importance and is the focus of a major Australian study attempting to prevent relapse in these children.
Greater awareness of the late effects of chemotherapy has led to changes in the treatment protocols for ALL, with improvement in neurocognitive outcomes and reduced rates of second malignancies.
Pharmacogenetics is a new field of research that aims to enhance treatment efficacy by assessing the individual’s metabolism of and response to chemotherapeutic agents.
Targeted therapies currently being developed show some promise of being able to further improve cure rates.
Adolescents with ALL have a better prognosis if treated with paediatric rather than adult protocols.